
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Associate professor, SSM College of Pharmacy, Jambai, Erode, Tamil Nadu, India. Pin code: 638311
2Pharm D, SSM College of Pharmacy, Jambai, Erode, Tamil Nadu, India. Pin code: 638311
Gaucher disease, pronounced "go-shey," is an inherited lysosomal storage metabolic disorders (LSDs) brought on by mutations in the glucocerebrosidase (GBA1) gene. A comprehensive search of relevant literature was conducted to identify relevant studies. Various electronic databases can be utilized. Keywords and search terms related to gaucher disease, early prediction, diagnostic strategies, and relevant biomarkers or imaging techniques will be used to maximize the search coverage. For case series, patients were recruited from various sites of Tamil Nadu, to identify the detrimental psychological and emotional effects of disease on patients and their families, by direct interaction with the patients. The study also created dietary guidelines with the support of experienced dietician for Indians living with Gaucher Disease for the purpose of prevention of complications, and empower patients and their families to take control of their own wellness. Our systematic review indicates that early prediction can reduce Gaucher disease morbidity and mortality. A case series study in Tamil Nadu, involving 9 patients (5 males, 4 females), identified 67% with type 1 GD. Enlarged liver and spleen were observed in 15% and 13%, respectively. Notably, 55% had consanguine marriages. In conclusion, the systematic review of the study has revealed the pattern of presentations observed in Gaucher Disease patients of Tamil Nadu and provides a thorough overview of the current evidence to enable development of targeted counseling guidelines for patients suffering from Gaucher Disease and their families in Tamil Nadu.
Lysosomal storage disorders (LSDs) are a group of over 70 inherited metabolic conditions that are brought on by mistakes in the genes that produce lysosomal proteins[1]. Philippe Gaucher presented the first account of Gaucher illness in a medical thesis in 1882 [2]. Mandelbaum classified the illness as systemic in the early 1900s[3]. It was determined in 1965 that the disease was lysosomal and lacked the enzyme acid glucosidase [4]. Gaucher disease, pronounced "go-shey," is an inherited metabolic disorder brought on by mutations in the glucocerebrosidase (GBA1) gene[5]. Three main forms of GD have historically been distinguished. Visceral symptoms mostly define Type 1 GD (GD1). The presence of splenomegaly, hepatomegaly, thrombocytopenia, anaemia, bone disease, and weariness are signs. There are neurological symptoms in both type 2 (GD2) and type 3 (GD3) illness, ranging from a more moderate neurological phenotype in GD3 to a rapidly deteriorating neurological state that results in mortality in the early years of life in GD2 [6].
Table-1 (Clinical Manifestations ) [7] [8]
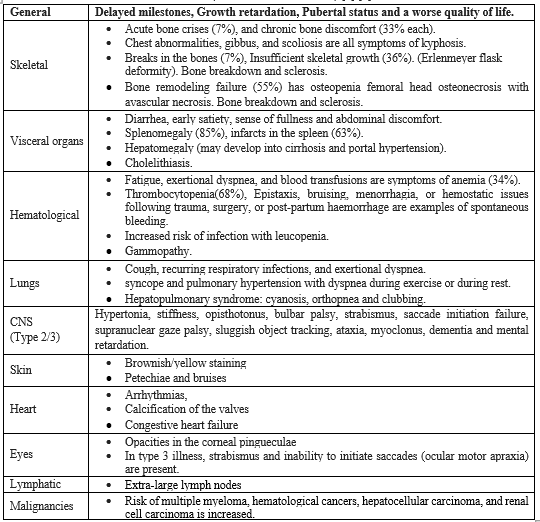
Prenatal genetic diagnosis of GD can be performed using either chorionic villus sampling (sampled at 10–12 weeks of amenorrhea) or amniotic fluid cells (as early as 16 weeks of amenorrhea) if the genotype of the index case has already been identified. On fresh chorionic villi or amniotic cell cultures, glucocerebrosidase activity may also be measured [9]. A 25% chance of recurrence in each pregnancy exists for GD, an autosomal recessive condition. Pre- and post-test counselling is necessary to make sure the family is aware of the risks associated with the condition throughout each pregnancy, the reproductive choices available to prevent recurrence, the consequences of giving birth to a child who has GD, and the alternatives open to them if the foetus is found to be afflicted. As of right now, all disease phenotypes in India consistently seek this as the most significant and successful method of reducing the burden of illness. Confirmation of the diagnosis in the afflicted child by enzyme testing and, preferably, also mutation identification, is a requirement for prenatal testing. Chorionic villus sampling is often carried out between 11 and 13 weeks of gestation. Conventionally, chorionic villi tissue is used to assess enzyme activity during prenatal foetal testing. If the family arrives late in the pregnancy, cultured amniotic fluid cells (15–18 weeks) or cord blood (19–20 weeks) can also be employed. If mutation studies are available, it would be ideal if biochemical data could be connected with them. It is crucial to be aware that any prenatal testing protocols should avoid maternal tissue contamination [10] [11]. According to the Medical Termination of Pregnancy Act, the family has the option to end the pregnancy up to 20 weeks of gestation if the foetus is afflicted. The method of choice for carrier identification is also molecular testing. Extended family members should be given it, especially in areas where consanguinity is prevalent. Experience with lysosomal storage disease prenatal diagnosis in India, the significance of proband diagnostic confirmation, and the detection of family variants have all been previously described [12] [13]. Siblings should be examined when a child in a family is diagnosed with Gaucher illness. Those who are discovered to be impacted but not yet exhibiting symptoms should be watched to determine any progression's rate. Even within siblings, there is significant phenotypic heterogeneity, therefore a presymptomatic diagnosis does not automatically indicate the need for treatment. These children should be checked for the onset of cytopenia, splenomegaly, and skeletal illness every six months [14]. ERT and SRT are effective treatments for GD in lowering or normalising liver volume. Alkaline phosphatase and aminotransferases in particular may be raised in GD patients' liver enzyme levels. However, the rise in liver enzymes is often minor or moderate and does not correspond to hepatomegaly or the degree of liver involvement [15]. Enzyme replacement therapy (ERT) is utilized as a first-line treatment for the condition. Eliglustat is a better oral therapy than ERT in terms of therapy time, compatibility with job, family, and social duties, and, most likely, improved quality of life [16]. Since the enzyme does not sufficiently cross the blood-brain barrier to be therapeutically effective, the CNS is not treated even while ERT safely improves liver, spleen, bone marrow, and harmatological sickness. SRT and other widely available substances that block glucosylceramide synthase may affect brain function and glucosylcemide levels, but they have not yet shown to be effective in correcting the neurologic phenotype [10]. About 50 cases of neuronopathic and non-neuronopathic GD have been treated with hematopoietic stem cell transplantation (HSCT). Stem cell transplantation is not currently a common procedure due to the accessibility of ERT and its favourable safety and effectiveness profiles. Our study encompass analyzing the distribution patterns of signs and symptoms among Gaucher Disease patients, developing tailored counseling guidelines for Gaucher Disease patients in Tamil Nadu, predicting early warning signs of the disease, assessing the utilization of prenatal screening for earlier pattern detection, and delivering customized counseling guidelines to assist the population of Tamil Nadu.
Fig 1 – Early Prediction Of Gaucher Disease
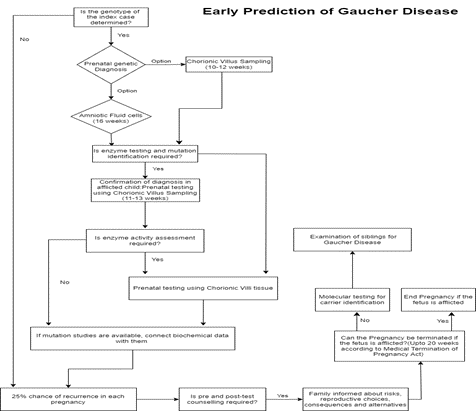
METHODS:
SYSTEMATIC REVIEW:
A comprehensive search of relevant literature will be conducted to identify relevant studies. Various electronic databases can be utilized. Keywords and search terms related to gaucher disease, early prediction, diagnostic strategies, and relevant biomarkers or imaging techniques will be used to maximize the search coverage. The search strategy may be developed with the assistance of a medical librarian or information specialist to ensure thoroughness and accuracy.
Fig 2 - Procedure for Systematic Review
INCLUSION CRITERIA:
We include relevant electronic databases like,
STATISTICAL ANALYSIS:
To find the mode of the systematic review of evidences.
CASE SERIES:
PARTICIPANTS:
DATA:
The following data was collected:
Demographic Information:
Age, sex and family history of the patient.
Clinical Presentation:
Symptoms, duration of symptoms, severity of symptoms, and organ involvement.
Diagnostic Information:
Results of biochemical and/or genetic testing, subtype of Gaucher Disease, and age at diagnosis.
Treatment: Type of treatment received.
STUDY CRITERIA
Inclusion criteria:
Exclusion criteria:
STUDY DESIGN:
OBTAINING CONSENT FROM THE ETHICAL COMMITTEE:
STATISTICAL ANALYSIS:
PATIENT COUNSELLING:
STATISTICAL ANALYSIS:
To find the outcome of patient counselling using TWO WAY ANOVA Test.
RESULTS:
Systematic Review
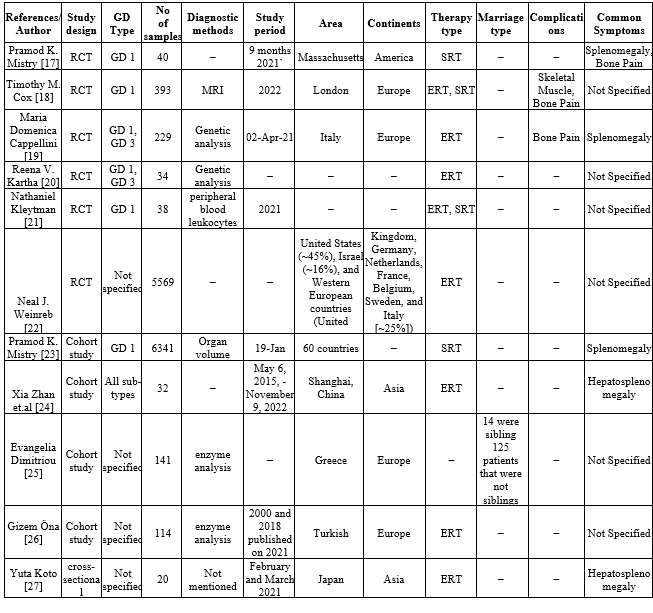
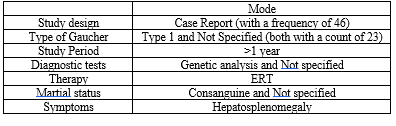
We compared the research articles on the disease from reliable databases like Pubmed and Embase, and we tabulated details like the author of the article, they employed a study design, the kinds of GD patients they saw in their study, The quantity of samples used, the preferred diagnostic techniques, Study time, study location, therapy methods, Marrital status of the parents, GD complications and the symptoms that accompany them.
Table-2 (Systematic Review of Evidences)
Study Designs
We noticed that they employed various study designs for their research when we analysed the systematic review of the evidence using pertinent databases. The majority of pertinent material came from case reports. A total of 75?se reports, 7?se series, retrospective studies, 4% cohort studies, and 1.5% cross-sectional studies were used. 1.5% of articles omit study design information.
Fig 3 – Study Designs used in Systematic Review of Evidences
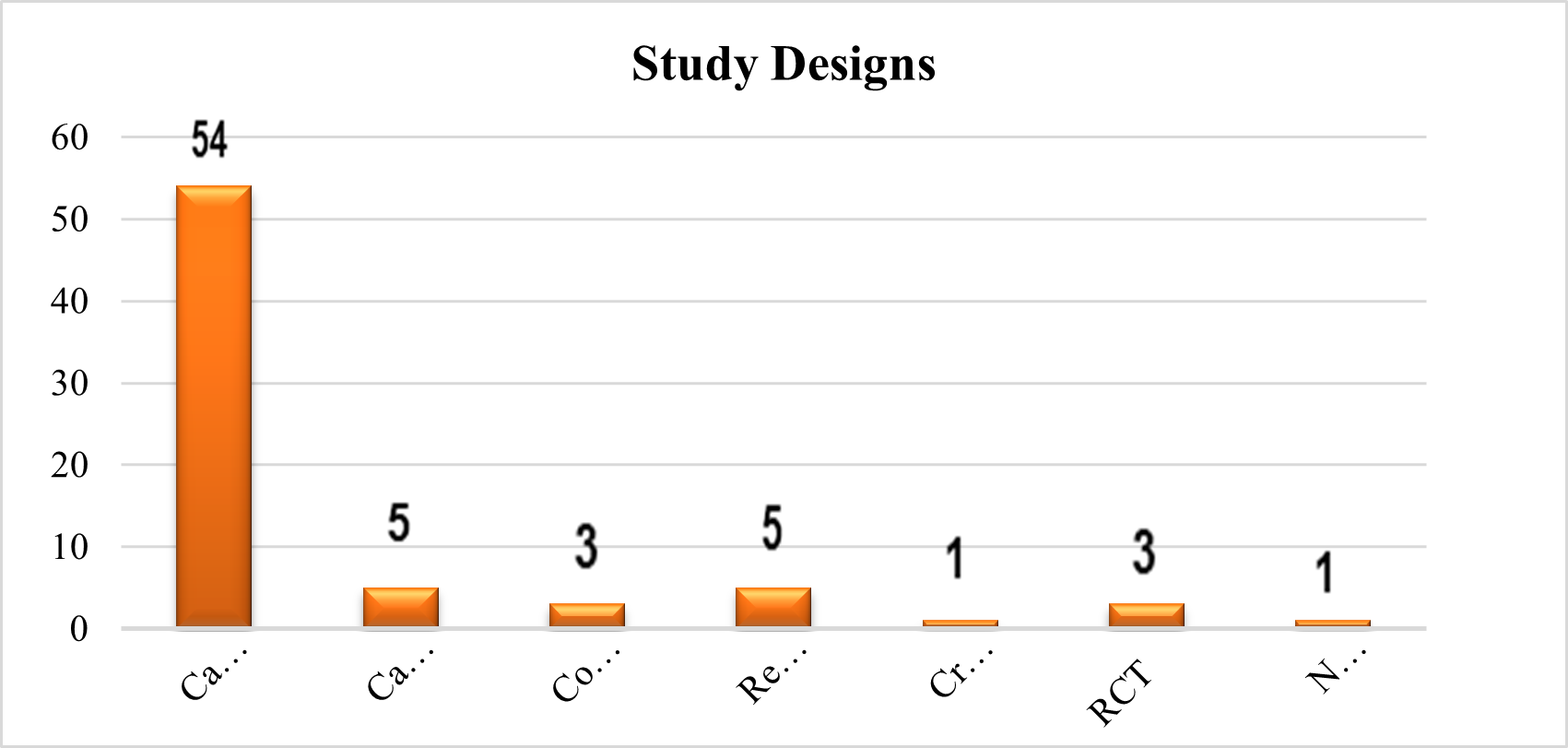
Types of GD Patients
Based on an analysis of the type-wise distribution of GD patients, it was discovered that 36 (50%) of them preferred type 1 individuals, 7 (10%) of them observed type 3 patients, 4 (6%) of them saw both type 1 and type 3, and 1% of them observed both type 2,3 and type 1,2,3. Patients with Type 2 GD were not observed. 23 (32%) of them did not specify the categories of patients with GD.
Fig 4 – Types of GD Patients used in Systematic Review of Evidences
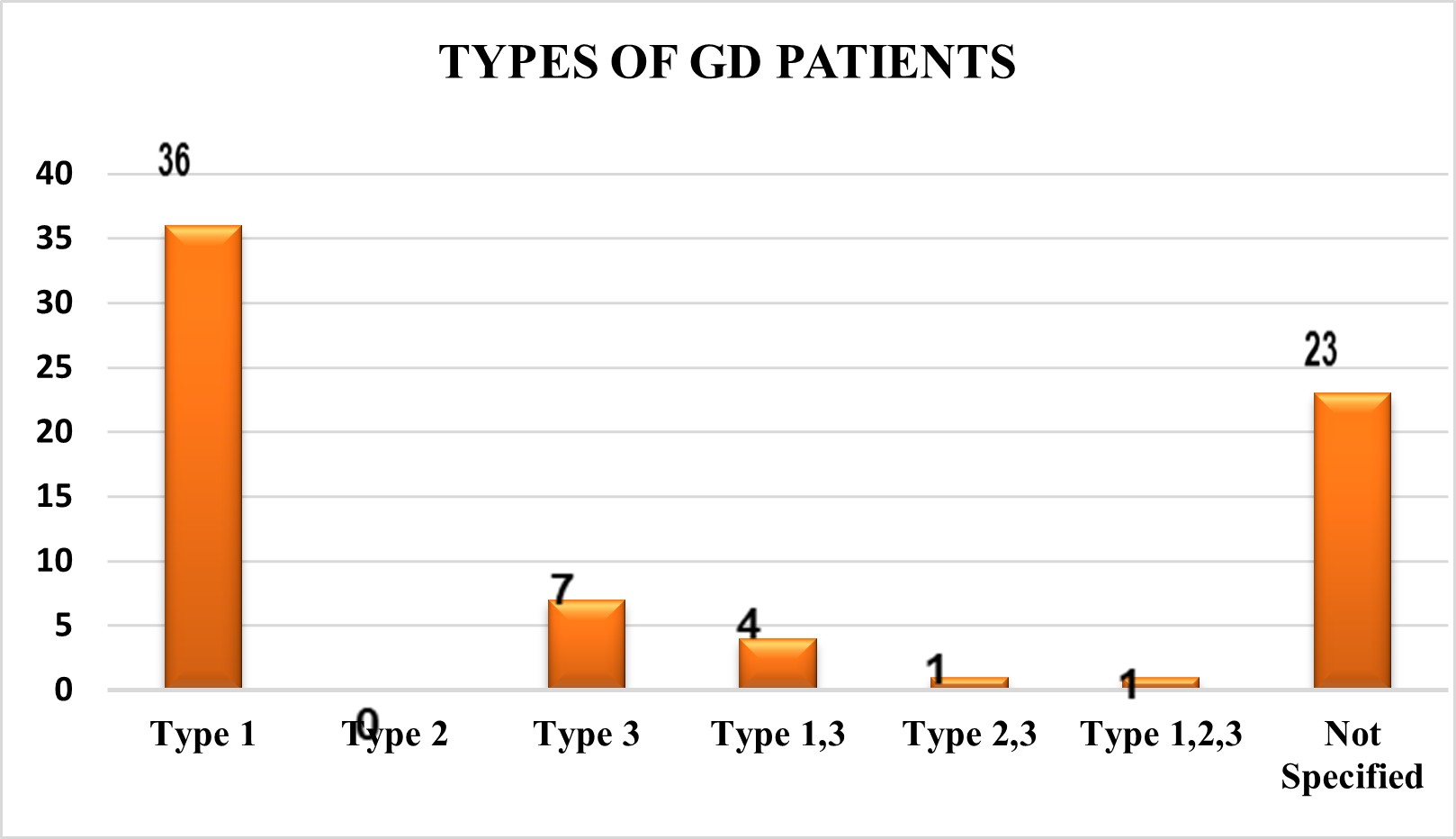
Study Period
In the systematic assessment of the data, most of the researchers 47% of them favoured study durations of less than one year, while 4% of them selected study durations of one to five years, five to ten years, and 2% of them favoured more than ten years. 43% of them failed to indicate the study's duration in their articles.
Fig 5 – Study Duration used in Systematic Review of Evidences
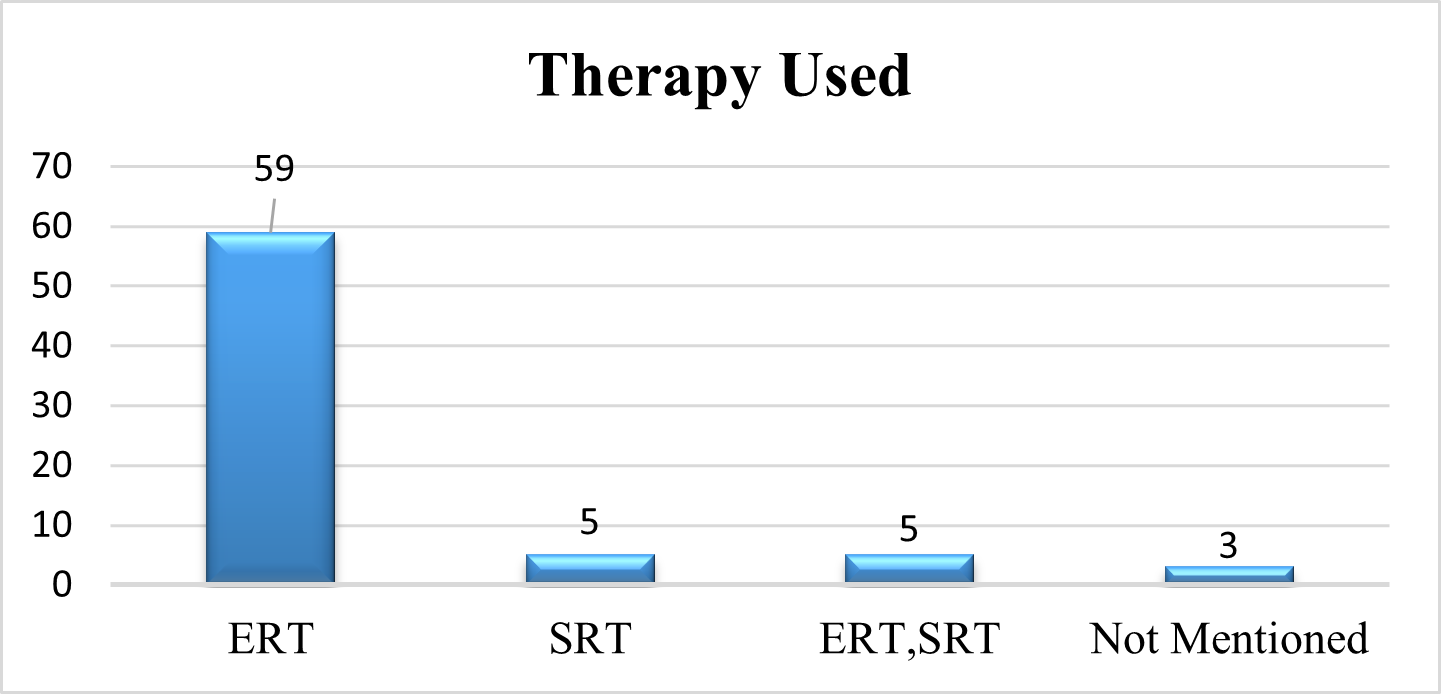
Therapy Used In GD Patients
As we examine the therapy use in patients with Gaucher disease using a systematic evaluation of the evidence. 82% of patients receive ERT, 7% of patients receive ERT, and 7% of patients receive both ERT and SRT. Three publications (4%) fail to disclose the patients' therapies.
Fig 6 – Therapy used in Systematic Review of Evidences
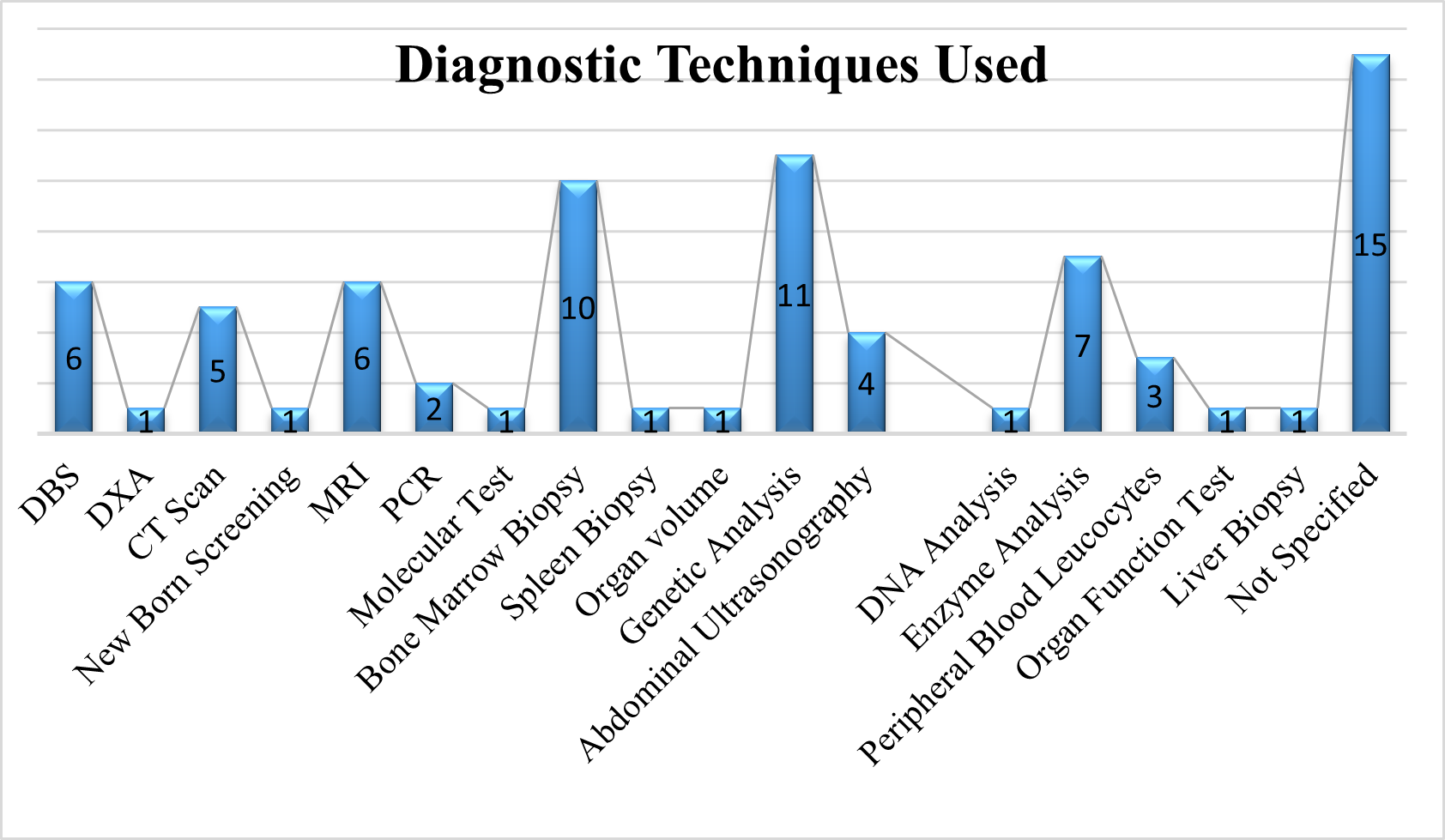
Diagnostic Techniques Used
As we analyse the diagnostic procedures used on GD patients using a systematic review of the evidence, 11 of them prefer genetic analysis, 10 prefer bone marrow biopsy, 7 prefer enzyme analysis, 6 prefer MRI and DBS, 5 prefer CT scan, 4 prefer abdominal scan, 3 prefer peripheral blood leucocytes, 2 prefer PCR, and 1 prefer DXA, newborn screening, molecular test, spleen biopsy, organ volume, DNA analysis, organ function test and liver biopsy
Fig 7 – Diagnostic Tests used in Systematic Review of Evidences
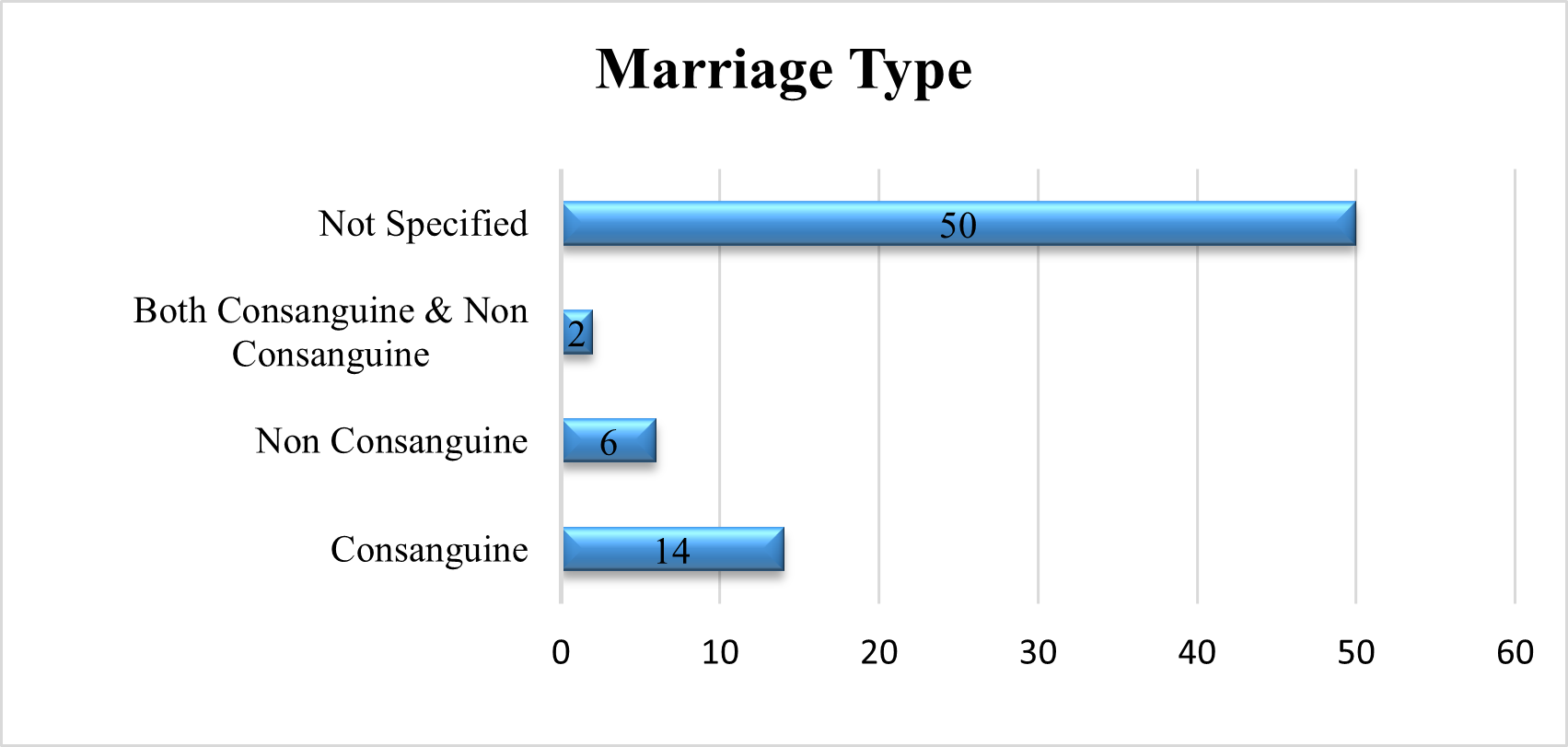
Marital Status Of Parents
In a comprehensive review of the literature, we evaluated the marriage status of the parents of GD patients. In 20% of the studies, they were married consanguineously, in 8%, they were not, and in 3%, they were married both consanguineously and not. In 69% of the studies, the parents' marital status was not disclosed.
Fig 8 – Marital Type of parents used in Systematic Review of Evidences
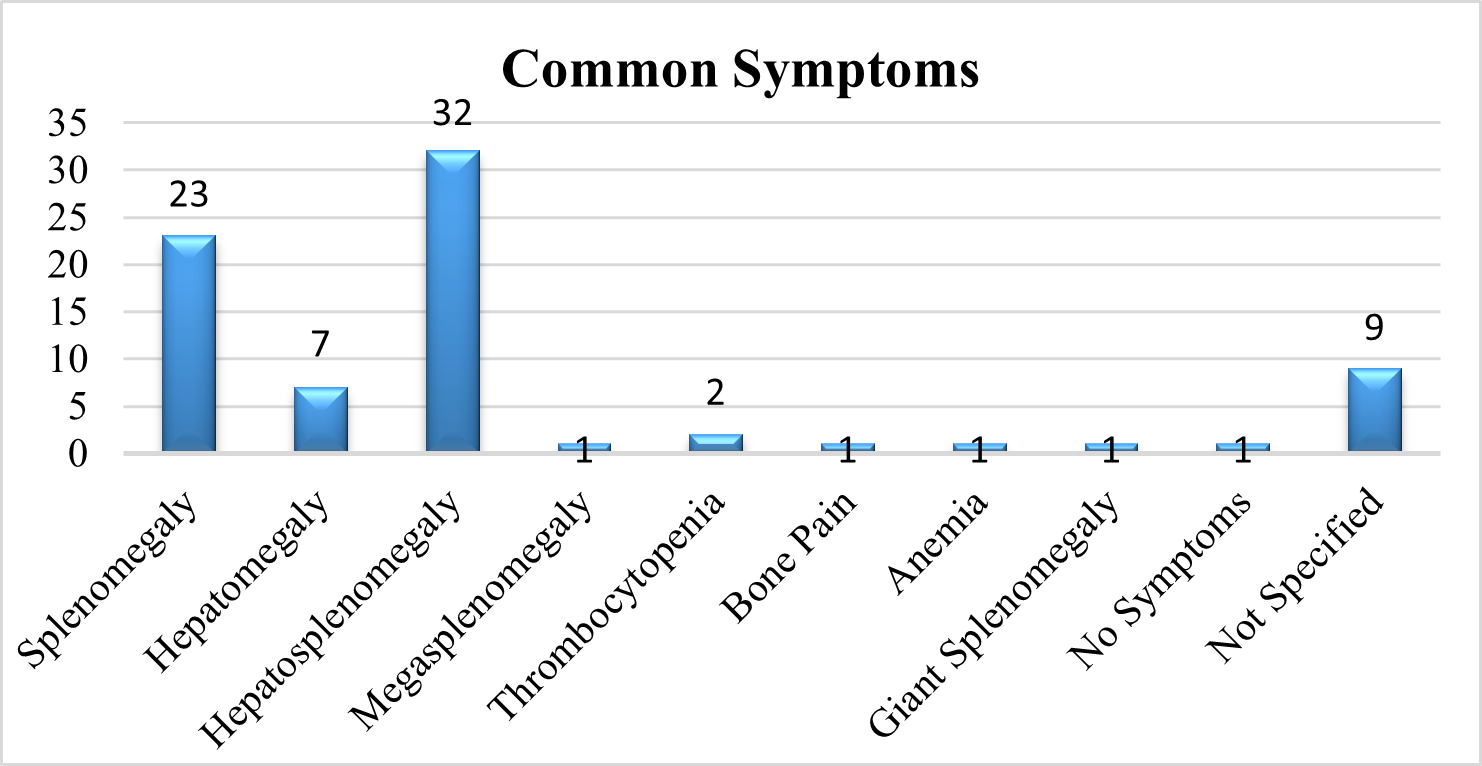
Common Symptoms Of Patient:
In a comprehensive review of the literature, we evaluated the symptoms of GD Patients. 41 % of the studies shows Hepatosplenomegaly, 30% of the studies show Splenomegaly, 9 % of the studies show Hepatomegaly, 3 % show Thrombocytopenia, whereas 1 % show Megasplenomegaly, Bone pain, Anemia, Giant splenomegaly and also with no symptoms. In 12% of the studies, symptoms of the patient was not disclosed.
Fig 9 – Common symptoms pf patients seen in Systematic Review of Evidences
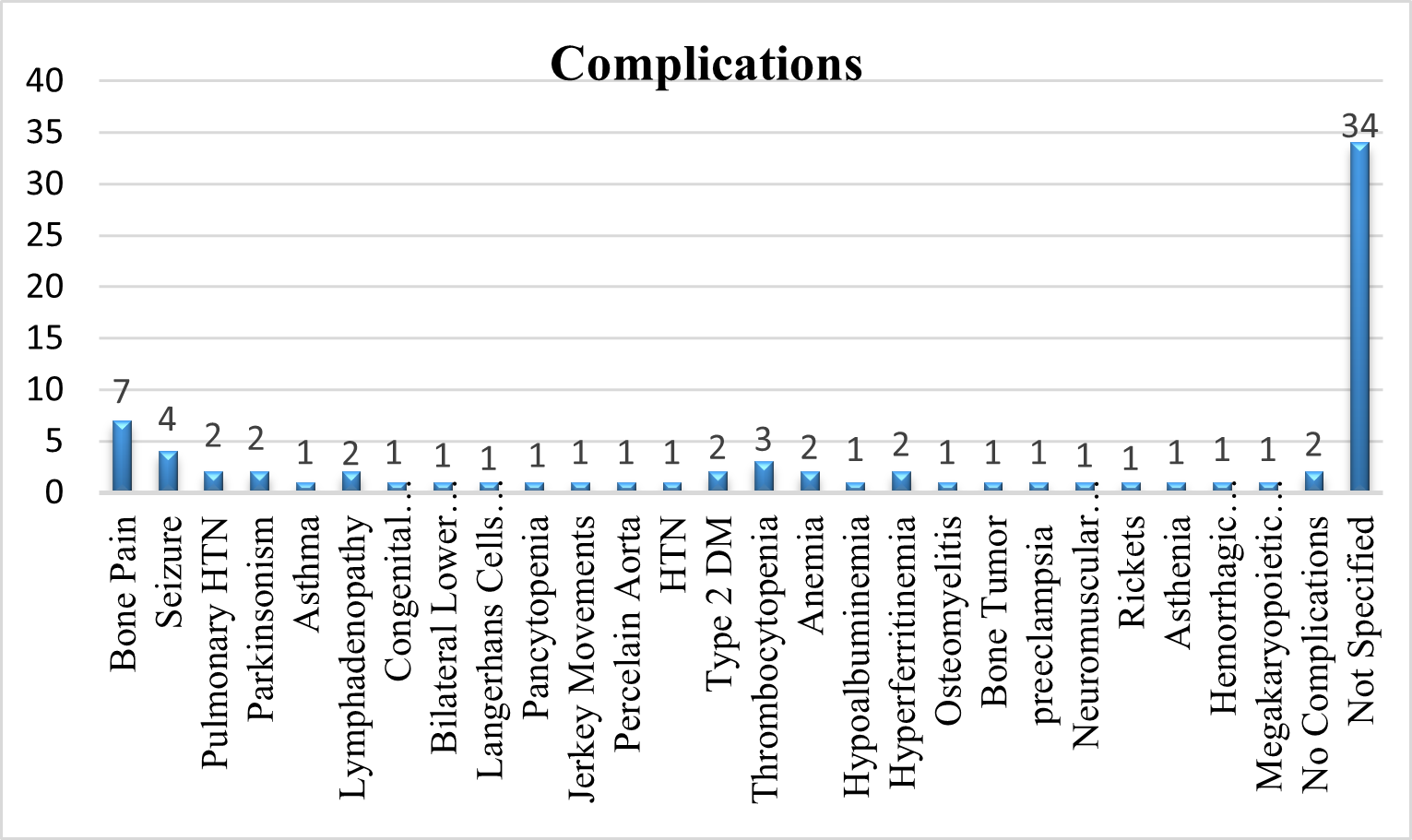
Complications
Complications of GD Patients observed in Systematic review was analyzed and reported.
Fig 10 – Complications of GD seen in Systematic Review of Evidences
STATISTICAL ANALYSIS OF SYSTEMATIC REVIEW:
Table-3 (Statistical Analysis of Systematic Review)
CASE SERIES
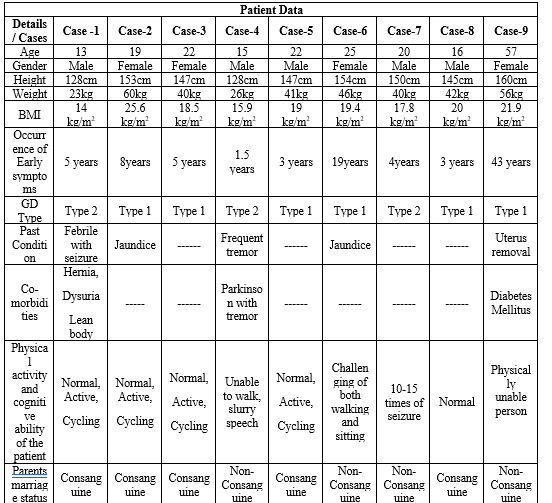
Patient Data:
Nine participants were chosen for our study. Eight of the patients in this group are between the ages of 13 and 25, while one patient is older than 50. As we analyzed the nine cases we tabulated the patient's characteristics, including when the symptoms first appear, their past medical condition, any comorbid conditions, GD types and cognitive and physical abilities.
Table-4 (Patient Data)
STATISTICAL ANALYSIS OF OUR CURRENT CASE SERIES:
Table-5 (Statistical Analysis of Case Series)

Case-1
A 13 year young male patient who was diagnosed with Gaucher Disease Type-2 when he was just five years four months old. He was hospitalized after expressing concerns about a fever, an enlarger stomach, respiratory issues, and abdominal pain. He had previously experienced a febrile seizure. He had a high temperature, rolling eyes, and jerking B/L upper and lower limbs at the time of admission. He was transferred to JIPMER hospital in Pondicherry where he received treatment for the aforementioned symptoms. The doctor also recommended a CT scan, which revealed that he also had an enlarged liver and spleen. They believed it was gaucher. As a result, the doctor advised taking a bone marrow test and their no further treatment was given. After consulting with Mediscan, they were advised to visit the Egmore VHS (Voluntary Health Services) children's hospital. Based on the comfort of the patient, they moved from here to Rajeev Gandhi Government Hospital in Chennai to Fortis Hospital in Vadapalani. After five years from the date of diagnosis, they were given an IV injection of 400U of imiglucerase. He is still taking the medication. In addition to the condition, he also had a hernia, dysuria, irregular growth, and a lean physique. Although his two siblings (one brother and one sister) were not diagnosed with Gaucher disease but, his brother was under suspicious of symptoms. The parents of the patient was not undergone any genetic examinations and their marriage was known consanguine. Their child both physical activity and cognitive ability was normal. The patient's parents were related via consanguineous marriage. The parents were a carrier of disease to their three childrens
Case-2
A 19 year young female patient who was diagnosed with Gaucher Disease Type-1 when she was eight years old. At the age of 7, she had a 20-day history of jaundice. Her symptoms upon admission were fever, cough, enlarged belly, difficulty breathing, abdominal pain, and yellow spots in her eyes. She was admitted with a high temperature and
a bloody cough. She was transferred to JIPMER hospital in Pondicherry where she received treatment for the aforementioned symptoms as well as a recommendation from the doctor to do a CT scan, which revealed that she had enlarged liver and spleen. They had a suspicion of Gaucher disease, therefore the doctor advised them to do an enzyme test; otherwise, and their no further treatment was given. They seek advice from Mediscan. Inj. Imiglucerase 30U IV Vial was administered to them 10 years after the diagnosis. She is still taking the medication. Other than the disease, she had no comorbidities. Her only sibling (brother) was not found to have gaucher disease. The parents of the patient was not undergone any genetic examinations and their marriage was non-consanguine. Their child both physical activity and cognitive ability was normal. The mother and father of both parents of the patient were the carrier of the disease. The patient father's side grandparents had eight children-four females and four males—while the patient mother's side grandparents had five female offspring.
Case-3
A 22 year young female patient who was diagnosed with Gaucher Disease Type-1 when she was five years old. She complained of a fever and an enlarged tummy when she was hospitalized. She didn't have any known past medical history. She was really tired and had a high temperature at the time of admission. She was treated for the aforementioned symptoms at Rajamurali Hospital, Sai Hospital in Thirumanavaayil, Child Trust Hospital in Nungampakkam, MIOT International Hospital in Porur, and finally Chettinad Hospital in Kelambakkam. The doctor also suggested a CT scan, and the results revealed that he had enlarged liver and spleen. They believed it was gaucher. In order to treat the patient further and remove the spleen due to massive splenomegaly (3kg), the doctor advised taking a bone marrow test. After consulting with Mediscan, they were advised to visit the Egmore VHS (Voluntary Health Services) children's hospital. After sixteen years from the date of diagnosis, they were given injections of imglucernase (3 vials/200 ml NS). She is still taking the medication. She had the comorbidities like easy occurrence of cough due to spleen removal. Her sibling (one sister) were not diagnosed with Gaucher disease. The parents of the patient was not undergone any genetic examinations and their marriage was known consanguine. Their child both physical activity and cognitive ability was normal. Two boys and three girls out of the five children that the patient's great-grandparents had were the disease-carriers. Their first male child had two sons and five daughters, their second had three sons and two daughters, their third had a daughter and a son, their fourth had three daughters and three sons, and their fifth had a son and two daughters.
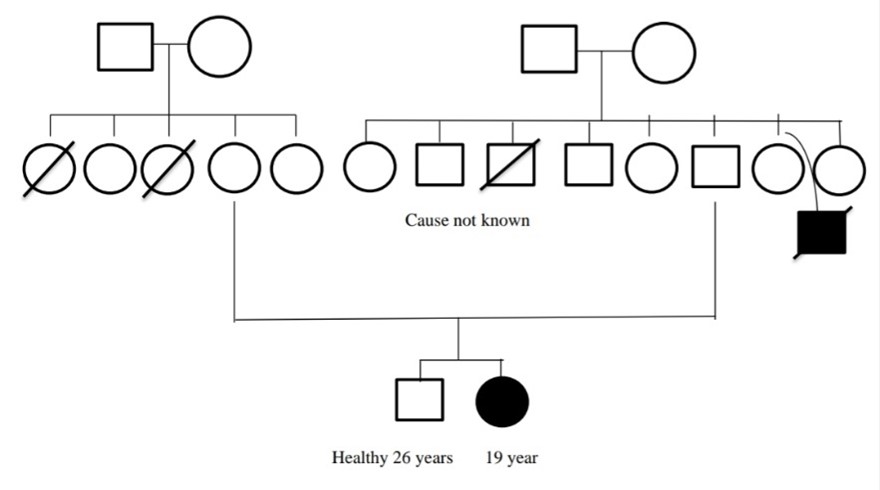
Case-4
A 15 year young male patient who was diagnosed with Gaucher Disease Type-2 at the age of one year six months. He was hospitalized after complaining of regular tremors, an enlarged stomach, acute exhaustion, and behavioral changes. His continuous multifocal MJ (+ history was recognized. At the time of admission he was extreme tiredness, enlarged belly and had frequent tremor. He was transported to Chennai Egmore hospital first, and subsequently to the Government hospital at Sengalpattu. He underwent a CT scan there at the doctor's recommendation, and the results revealed an enlarged liver and spleen. They believed it was gaucher. So, the doctor advised taking a bone marrow test, and a Mediscan revealed the disease. After seven years from the date of the diagnosis, they were given Inj. Imiglucerase 30 U IV Vial. He is still taking the medication. He had a slight improvement in his health while receiving therapy, such as a reduction in the enlargement of his liver and spleen. He had comorbid conditions including Parkinson's disease and tremor. Since the onset of his tremor at age 10, he has been unable to walk for a while till now. His sibling (one brother) were not diagnosed with gaucher disease. The parents of the patient was not undergone any genetic examinations and their marriage was non-consanguine. Their child had abnormalities in both physical activity and cognitive capacity, such as difficulty walking or speaking clearly (slurred speech), and he was confined to lying down on the bedside due to frequent tremors.
Case- 5
A 22 year young male patient who was diagnosed with Gaucher Disease Type-1 at the age of 3. His symptoms of a bloated stomach, anemia, being unable to eat, and bone weakness led to his admission. He didn't have any recognized medical history. He was transferred to KMCH Hospital in Coimbatore where he received treatment for the aforementioned symptoms. The doctor also recommended a CT scan, and the results revealed that he had an enlarged liver. They suspected for Neeman Pig Disease on the brain. The therapy didn't get any better after the parents took him to get siddha drugs. Following their consultation with Mediscan, the disease was confirmed. After five years from the date of diagnosis, they were given six vials of imiglucerase in 300 ml NS over the course of 90 minutes. He is still taking the medication. Additionally, they took Nasal spray 1 puff of 0.5 mg if the seizure lasted longer than two minutes and Tab. Levetiracetam 500 mg BD for two months. Beyond the disease, he had no comorbidities. Gaucher disease was not identified in his siblings. The patient's parents did not undertake any genetic testing, they were not consanguineous, and a member of his family passed away before the individual turned five. Both the child's physical and mental development were normal. Five boys and three girls out of the eight children that the patient's great-grandparents had were the disease-carriers.
Case-6
A 25 year young female patient who was diagnosed with Gaucher Disease Type-1 at the age of 19. She was admitted after complaining of a fever, an enlarged belly, extreme exhaustion, abdominal pain, yellow spots in the eyes, intermittent eating, anemia, bone pain, digestive issues like vomiting, loose motion, and sleeping problems. She had a history of significant hip discomfort and jaundice when she was nine years old. She also underwent a Varus derotation confinement Osteotomy® proximal femur procedure. She had fainted at the time of admission due to anemia. After being transported to ESI Hospital in Salem, they were transferred to ESI Hospital in KK Nagar, Chennai, where they suspected a lymphoma case. The patient also had a history of intermittent fever. Patient complained of easy fatigability and loss of appetite. The patient was treated for the aforementioned symptoms for three months and the doctor advised a CT-Scan, which revealed that she had an enlarged liver and spleen in addition to widespread body discomfort with right hip pain. She had no prior history of bleeding. Gaucher disease was suspected, therefore the doctor advised more testing. A bone marrow biopsy was used to confirm the diagnosis, and after one year from the date of diagnosis, the patient was treated with injections of imiglucerase 4000U IV 7 Vial/200ml NS over two hours once every 15 days. She has continued to take the medication for the past three years, although the dosage has been decreased to 60 IU/kg once every 15 days. Additionally, she received a monthly dose of Tab. folic acid. Her health slightly improved as a result of the therapy, including a reduction in spleen swelling and an increase in blood volume. Patient had been discharged and sent to MMC in Chennai for additional assessment and treatment.
She had no comorbidities apart from the disease. Her sibling (one brother) was not diagnosed with Gaucher disease. The parents of the patient was not undergone any genetic examinations. She was a second child born to non-consanguine parents. Her child hood was normal except for jaundice. Their child physical abnormality was difficult to sit in the floor and walk in long distance due to pain in her right leg because of surgery. Cognitive ability of the patient was normal.
Case-7
A 20 year young male patient who was diagnosed with Gaucher Disease Type-2 at the age of 4. He was hospitalized after reporting to have a fever, an enlarged stomach, bone pain, dizziness, and a sleep disturbance. He had previously experienced a febrile seizure. He had a high temperature, rolling eyes, and jerking B/L upper and lower limbs at the time of admission. He was transferred to Apollo Hospital in Chennai, where Dr. Revathiraj treated him for the aforementioned symptoms. The doctor also recommended a CT scan, and the results revealed that he had an enlarged liver and spleen. They believed it was gaucher. Therefore, the doctor advised patients to contact Dr. Aba Nakral at Paatiya Hospital in Mumbai (SANOFI). After a year from the date of diagnosis, they got Inj. Imiglucerase 400U IV Vial from their doctor, Sujatha. He has been taking the medication up to this point, and it has improved his condition and his ability to walk. In addition, other medications have been given to him to alleviate the drug's negative effects. But the other drug was not mentioned He had no comorbidities apart from the disease. His sibling (one sister) was not diagnosed with Gaucher disease. The parents of the patient was not undergone any genetic examinations and their marriage was non-consanguine. Their child both physical activity and cognitive ability was abnormal like 10-15 episodes of seizure at day time and unable to speak clearly.
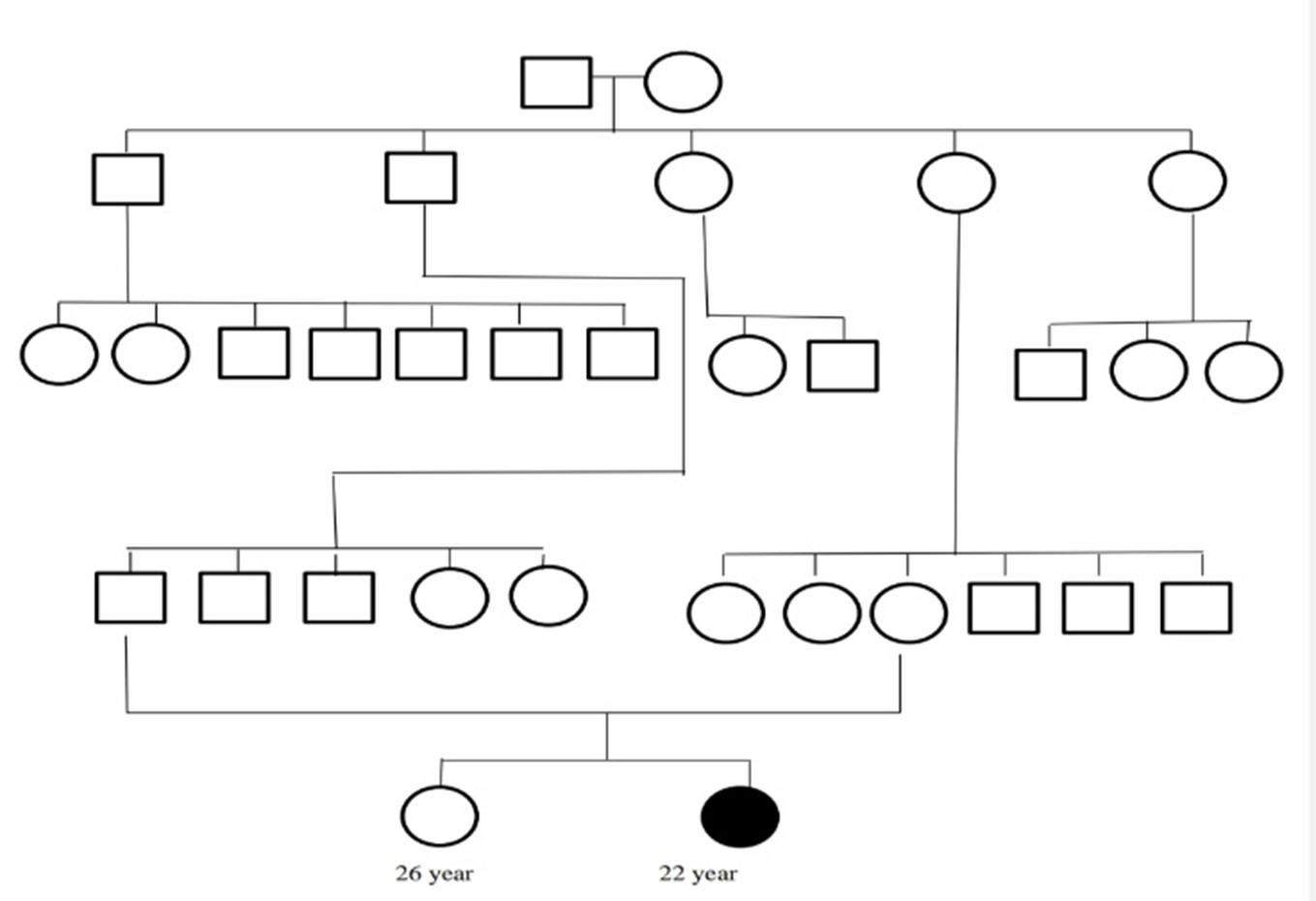
Case-8
A 16 year young male patient who was diagnosed with Gaucher Disease Type-1 at the age of 1. He was admitted after complaining of a headache, leg discomfort, and an enlarged tummy. He didn't have any recognized medical history. He was transferred to Apollo Hospital in Chennai where he received treatment for the aforementioned symptoms. The doctor also recommended a CT scan, and the results revealed that his spleen was enlarged. They believed it was gaucher. Then, after one year from the date of diagnosis, they received Inj. Imiglucerase 400U IV 3 Vials after the doctor advised them to consult with Mediscan. He continued taking the medication up until this point, and he was just dealing with the condition itself. He had no comorbidities apart from the disease. His sibling (one sister) was not diagnosed with Gaucher disease. The parents of the patient was not undergone any genetic examinations and their marriage was non-consanguine. Their child both physical activity and cognitive ability was normal. The child had physical abnormalities like eye problem
Case-9
A 57 year old female patient who was diagnosed with Gaucher Disease Type-1 at the age of 37. Her symptoms upon admission included an enlarged tummy, anemia, bone pain, dizziness, excessive fatigue, and weight loss. When she was 12 years old, she was diagnosed with bone tuberculosis, which caused her bone density to decline. Her uterus was removed in 2009. She was transferred to the government hospital in Madurai, where the doctor treated her for the aforementioned symptoms and recommended a CT scan, which revealed an enlarged liver. They suspected for gaucher. After that, they traveled to Vellore's CMC and subsequently to Chennai's Stanley Hospital. They obtained Inj. Imiglucerase 400U IV 6 Vials and she has been taking the medication up to this point after the doctor there advised them to confirm it with Mediscan. While using the medication, skin allergies and body aches were described as side effects, while sleepiness and exhaustion were observed without the medication. She had comorbidities like Diabetes Mellitus, Thrombocytopenia, and Anemia. Her siblings (two brothers and three sisters) were not diagnosed with Gaucher disease. The parents of the patient was not undergone any genetic examinations and their marriage was non-consanguine. The patient's physical activity was abnormal because she was too exhausted to work constantly; as a result, she slept, and her cognitive function was normal. She was born with anemia. She was unable to walk correctly due to physical defects like osteoarthritis.
SYMBOLS:
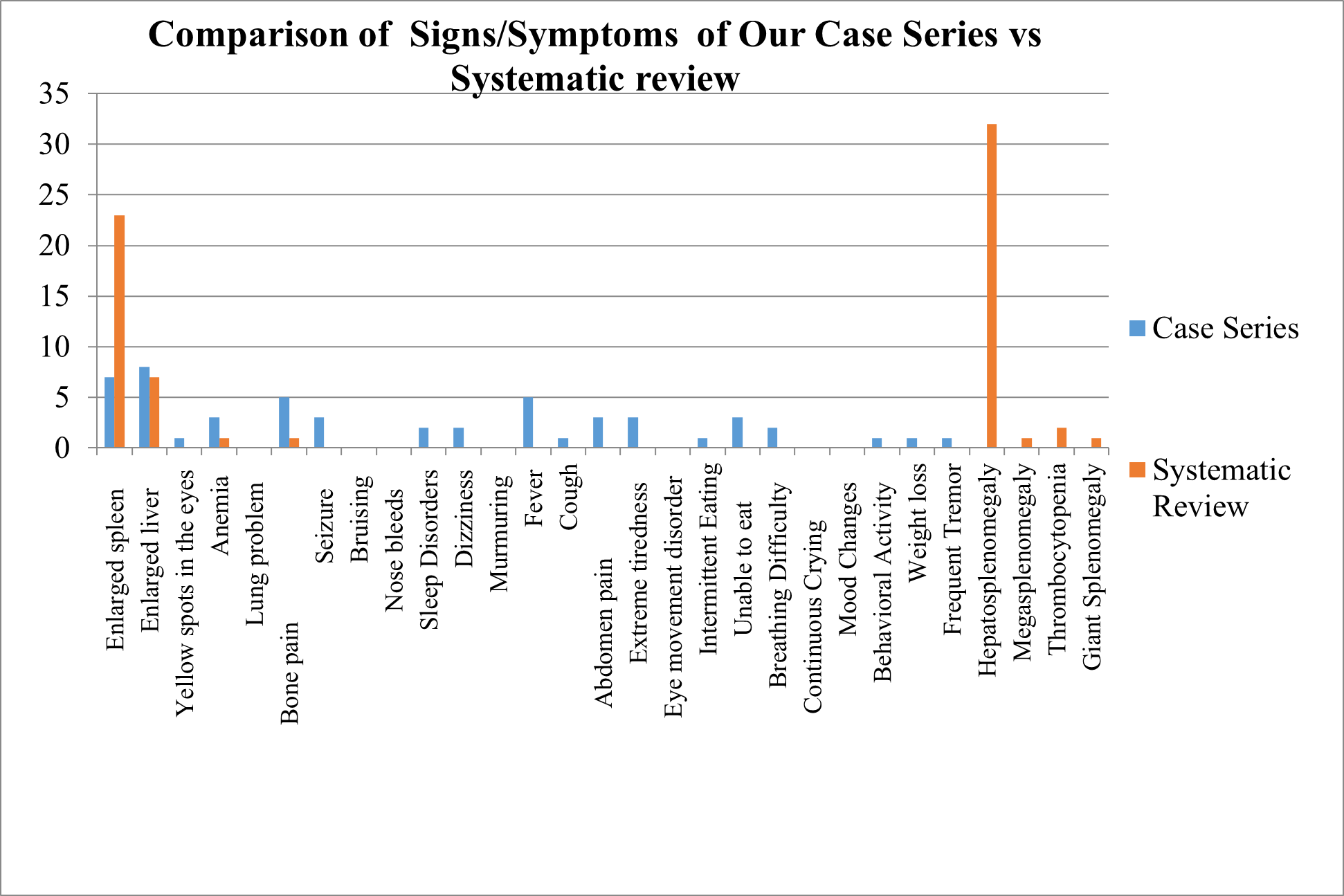
OUR CURRENT CASE SERIES VS SYSTEMATIC REVIEW:
Comparison of Sign/Symptoms
We compared the sign and symptoms in GD Patients of Case series with Systematic review of evidences
Fig 11 – Comparison of Diagnostic Tests of Case Series vs Systematic Review
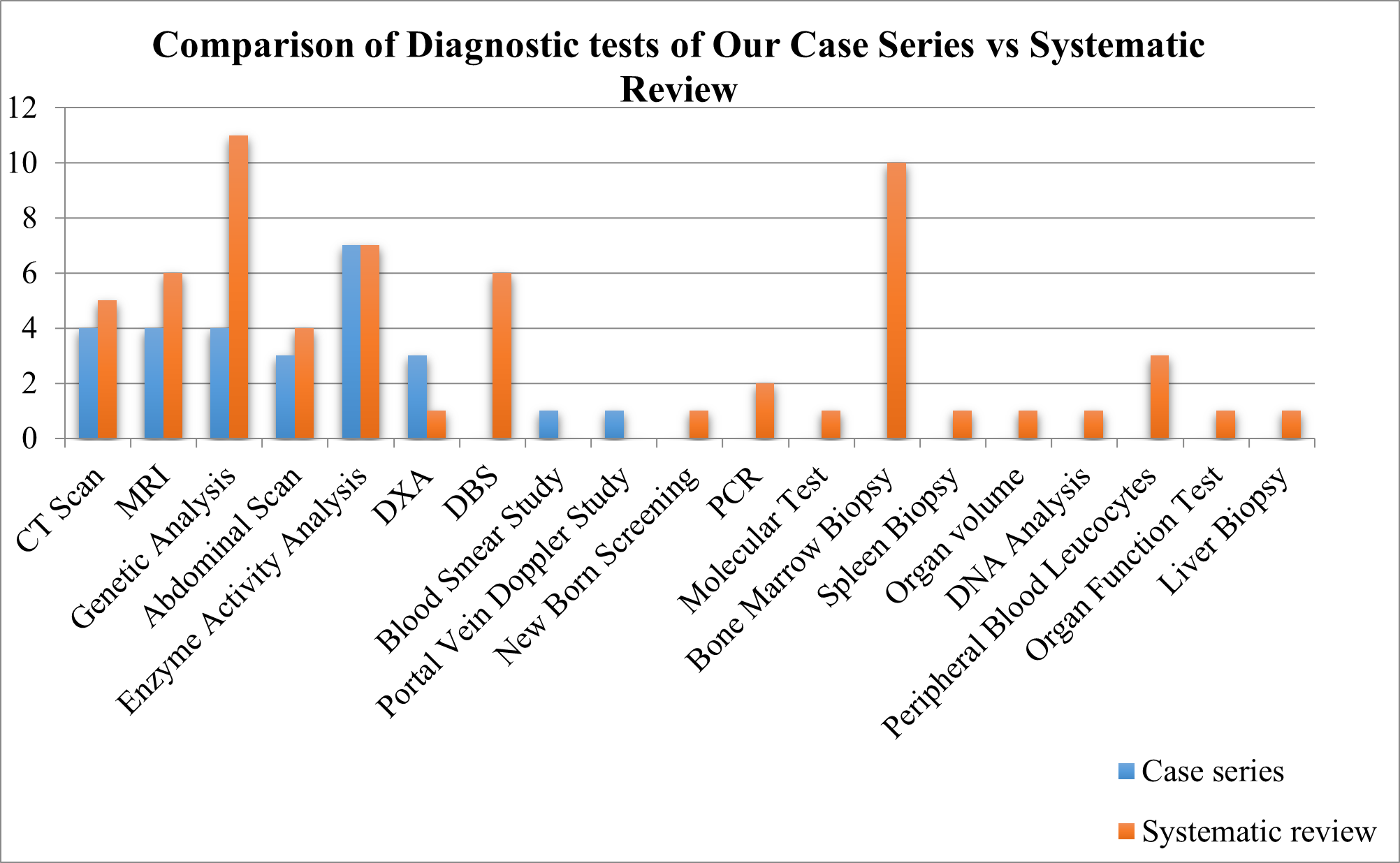
Comparison of Diagnostic Tests
We compared the Diagnostic Tests taken in GD Patients of Case series with Systematic review of evidences
Fig 12 – Comparison of Diagnostic Tests of Our Case Series vs Systematic Review
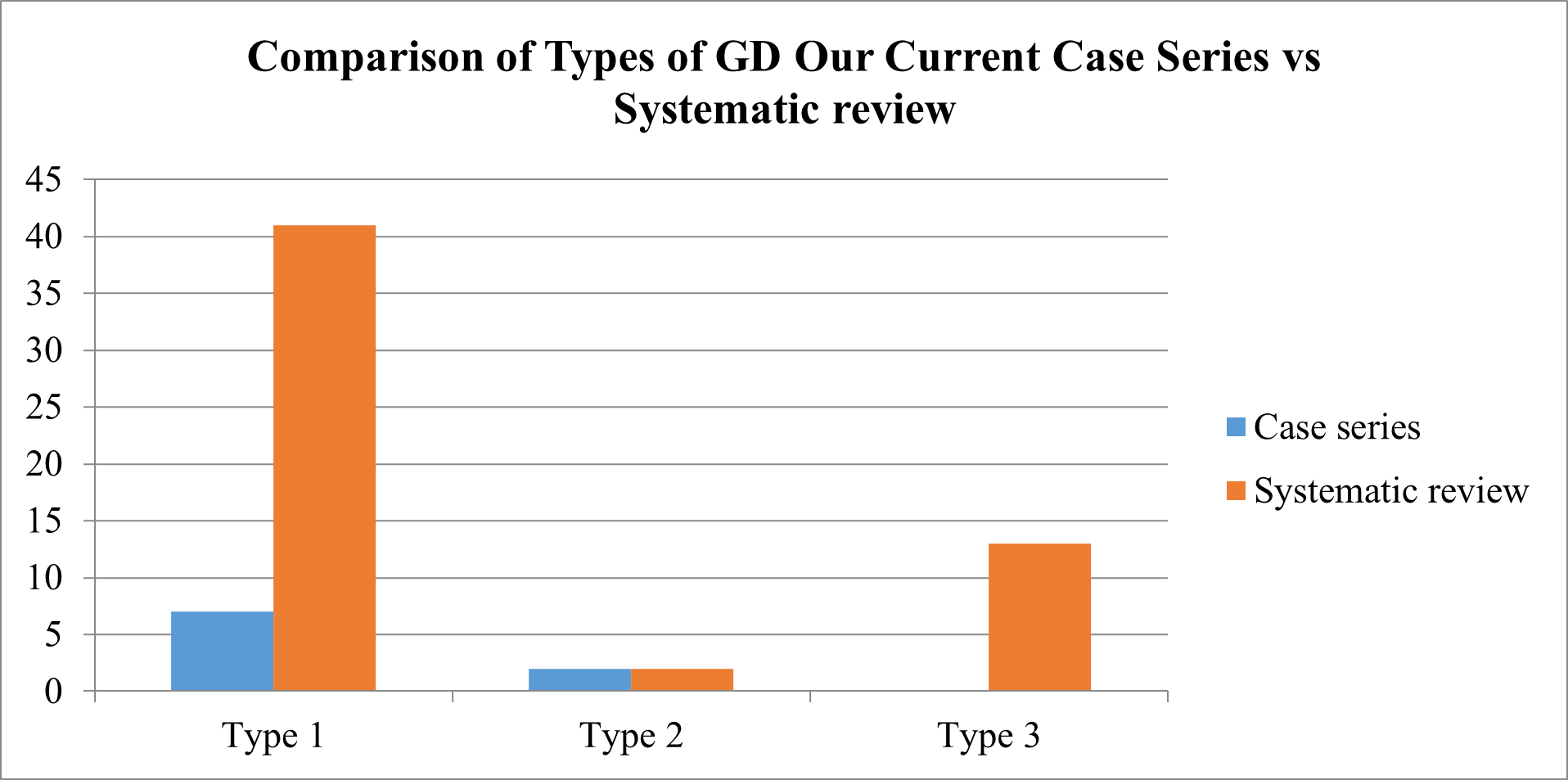
Comparison of GD Types
We compared the Types of GD Patients of Case series with Systematic review of evidences.
Fig 13 – Comparison of Types of GD patients of Case Series vs Systematic Review
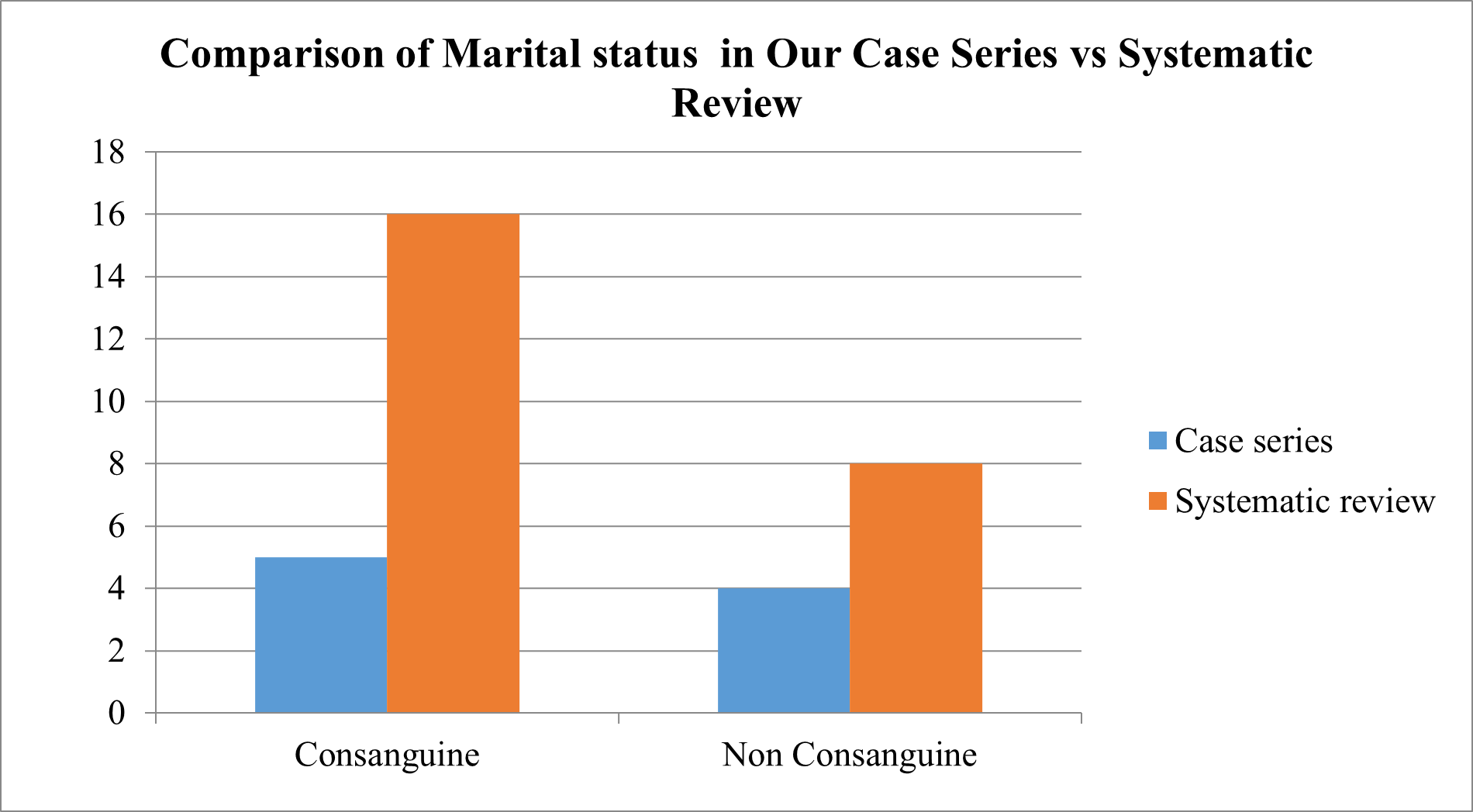
Comparison of Marital status of Parents of GD patients
We compared the Marital status of Parents of GD patients of Case series with Systematic review of evidences
Fig 14 – Comparison of Marital status of Parents of GD patients
PATIENT COUNSELLING RECOMMENDATIONS:
The following patient counselling recommendations were created for Tamil Nadu gaucher disease patients or their families.
Genetic Counselling:
A crucial component of the treatment provided to these individuals and their families is genetic counselling. As the child's condition worsens, parental sentiments of shame or blame might make decision-making more difficult. The dangers and options associated with various methods of family planning, such as adoption, sperm or egg donation, prenatal diagnosis, and pre-implantation diagnosis, need to be explained to families. The genetics team should also be aware that patients with GD have been characterised by atypical mechanisms such as uniparental disomy for chromosome 1 and germline or de novo mutations [89] [90]. Although no professional organisation recommends general population carrier screening for GD, the condition is included on clinically accessible pan-ethnic carrier screening panels, and 23% of genetic counsellors said they would provide GD carrier screening to any patient who requested all available prenatal screening tests. The prevalence of Gaucher disease may be higher in India due to the custom of consanguineous marriages in some areas of the nation. L444P seems to be the most common +-mutation for Gaucher disease in India out of the more than 300 mutations that have been recorded [91]. Siblings should be examined when a child in the family is diagnosed with Gaucher's illness. Those who are discovered to be impacted but not yet exhibiting symptoms should be watched to determine any progression rate. Even within siblings, there is significant phenotypic heterogeneity, therefore a pre-symptomatic diagnosis does not automatically indicate the need for treatment. These children should have their cytopenia, splenomegaly, and skeletal conditions checked every six months [92]. When necessary, suitable counselling and assistance should be suggested. Additionally, with the parent's permission, informing coworkers, relatives outside the immediate family, and clergy about the seriousness of the sickness can assist the family get the necessary support and quality time with the kid. It might also be highly beneficial to use hospice care[93]. The autosomal recessive nature of Gaucher disease (GD) makes it inheritable. Each afflicted person's sibs have a 25% risk of being affected at conception, a 50% chance of being asymptomatic carriers, and a 25% chance of being unaffected and not a carrier [94]. Therefore, supporting their generation rather than serving as a major means of avoiding Gaucher illness is the goal of genetic counselling. The panel favoured genetic counselling for Gaucher disease patients and their families. To identify additional people who may be at risk and to explain future reproductive possibilities, genetic counselling should involve a thorough family history and pedigree. It should also explain how genotype affects phenotype and how neuronopathic disease risk relates to it [95]. GD is an autosomal recessive condition, but difficulties in providing genetic counselling for an autosomal recessive inheritance include (1) a lack of family history; (2) occasionally, autosomal recessive conditions occur in sequential generations; and (3) if the biological father of an affected individual is someone other than the person assumed to be the father, it may lead to false carrier test results (the apparent father would typically not be a carrier) and risk of adding a. Given that there is a 3%–5?seline risk for a birth defect or genetic disorder, prenatal genetic counselling is pertinent to all pregnancies [96]. Therefore, we advised them to verify their siblings' prenatal screening results. Although no professional organisation advises broad population carrier screening for GD, GD is listed clinically. In addition, 23% of genetic counsellors indicated they would provide GD carrier screening to any patient who requested all prenatal screening tests, including the relevant pan-ethnic carrier screening panels [97].
Diet Based Counseling:
Drinks, dairy products and milk, poultry, eggs, meat, grains, vegetables, beans, fruit, and (sweets, fried meals, and fast food) were the several food categories that the goods fell under. The number of cups or glasses for coffee or soft drinks was also given[98]. Organomegaly-related gastrointestinal symptoms, including early satiety, abdominal bloating, and stomach heaviness, can occur in some GD patients. By breaking up meals into smaller and more frequent amounts and cutting back on food volume, those symptoms can be alleviated [99]. Individuals with GD should be provided bone health guidelines comparable to those given to people who have osteoporosis even if there have not been studies on the impact of food or lifestyle on the course of skeletal deterioration. These include getting enough vitamin D and calcium from food, as well as engaging in regular exercise. Malnutrition is another issue that some GD patients struggle with, and it can aggravate skeletal deterioration. An energy- and high-in protein diet is advised for these people [100]. Patients with GD have increased resting energy expenditure[101] [102] [103] and this may contribute to their growth delay and cause them to be underweight[104]. There is no specific diet that can treat or cure Gaucher's illness. However, some dietary modifications may aid in symptom management and improve general health. According to a specific study, the following nutritional condition exists. A low-fat diet is suggested to reduce the risk of liver disease and maintain a healthy weight. You may get the essential vitamins and minerals you need to maintain your overall health by consuming a range of fruits, vegetables, and whole grains. Some people with Gaucher disease may benefit from a high-calorie diet to maintain a healthy weight and prevent starving. To build a personalised nutrition plan that takes into account your unique requirements, you must work with a certified dietitian or other healthcare professional. Vitamins, minerals, and nutrients abound in fruits and vegetables. Antioxidants found in them also aid in lowering inflammatory and oxidative stress. Whole-grain foods are a great source of fibre, vitamins, and minerals. Brown rice and oats are two healthful grains that persons with the condition Gaucher can eat. Consuming lean proteins including chicken, turkey, seafood, legumes, and lentils is advised for those with Gaucher disease. Lean proteins are low in fats and can support good weight management. Inflammation in the body can be decreased by eating healthy fats such as the omega-3 fatty acids found in fish, nuts, and seeds. Osteoporosis can happen to people who have Gaucher disease. Dairy products, green leafy vegetables, and fortified meals are examples of foods high in vitamin D and calcium that can help maintain healthy bones. Dietary recommendations for Gaucher's illness should also be addressed by a doctor or a qualified dietician[105][106]. Although GD1 was formerly thought to be non-neuronopathic, it has now been shown that those who carry it are more susceptible to developing Parkinson's disease (PD), a prevalent neurodegenerative condition that affects around 2% of persons over the age of 80 [107]So, we saw a single person who had GD along with PD. We advised them to listen to our advice and do what we said.
OUTCOME OF PATIENT COUNSELLING:
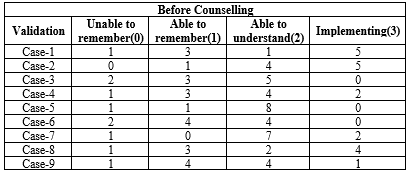
Nine patients underwent counseling, and after a month, follow-up phone calls were made. Ten questions, depending on the counselling provided, were asked to patients or their families, and the four responses—unable to recall, able to remember, able to understand, and implementing—were obtained from the patient. For the aforementioned responses, a score of 0,1,2,3 was provided.
OUTCOME OF BEFORE COUNSELLING BY US:
Table- 6 (Outcome of Before Counselling By Us)
OUTCOME OF AFTER COUNSELLING BY US:
Table- 7 (Outcome of After Counselling By Us)
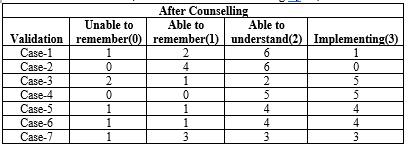
Table- 8 (P Value for TWO WAY ANOVA TEST)
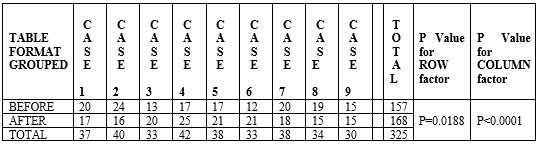
P Value for ROW factor (P=0.0188) is considered as statistically significant.
P Value for COLUMN factor (P<0>
Fig 15-Outcome of patient counselling in graph

DISCUSSION
Out of 1431 studies selected from databases like Pubmed/Medline, Science direct/Embase, we extract 72 studies based on our inclusion and exclusion criteria. There were 46 Case report studies, 9 Case series studies, 6 RCT studies, 5 Retrospective studies, 4 Cohort studies, 1 Cross sectional and Descriptive study among the study designs. These research looked at variety of topics, including peoples with GD Type, symptoms, early prediction possibilities, prevalence of GD, Diagnostic methods, marital type, complications and therapy type in patients with GD. Our study was compared to another study observed that Data extraction was done using 18 studies that were selected from 1874 records. The research that met the requirements for inclusion covered a total of 15 regions/countries. Globally, there were 1.5 instances of GD per 100,000 live births (95% confidence interval: 1.0 to 2.0). Globally, there were 0.9 instances of GD per 100,000 people (95% confidence interval: 0.7 to 1.1) [108]. The SF-36 was the instrument that was used the most frequently out of the 24 different patient-reported outcome assessments throughout the 33 studies that were found. There were 18 cross-sectional studies, 7 pre- and post-intervention studies, 3 randomised controlled trials, 2 cohort studies, 2 qualitative studies, and 1 validation study among the study designs. These research looked at a variety of topics, including people with GD's quality of life and cardinal symptoms such exhaustion, pain, bleeding, cognition, social interactions, and psychological functioning. Additionally, the evaluation of the therapeutic advantages of various therapies in patients with GD received significant attention. There has also been the development of a fresh GD-specific instrument with two practical applications: a 24-item variant for routine clinical monitoring and a 17-item form for use in clinical trials[109]. This study analyzes a case series of active individuals with Gaucher disease in Tamil Nadu, focusing on their struggles in managing the disease and improving their quality of life. Nine patients had gaucher disease with 8 in the liver, 7 in the spleen and 5 had fever and bone pain/weakness. CT Scan, MRI, Enzyme analysis and Genetic analysis revealed Gaucher Disease in patients. In this study 6 had Type 1 GD and remaining 3 had Type 2 GD. Our study was compared to another study observed that Seven adult patients had gaucheromas, with 4 in the spleen, 2 in the liver, and 1 in the bone. Both MRI and ultrasound examinations revealed no signs of cancer. Ten studies in the literature described Gaucheromas, the majority of which were found in the liver and spleen in people with type 1 GD. All of our patients had type 1 GD, and during the five years of follow-up, the ultrasonography aspect remained unchanged[39]. For a comparative analysis of systematic review and case series on Gaucher’s Disease (GD), we explored multiple facets of their research conclusions. We identified several commonalities with respect to warning signals and manifestations. Systematic reviews suggested a more extensive incidence of hepatosplenomegaly but, at contrast, case series indicated an increased presence of enlarged liver signs or symptoms. Moving on with diagnostic tests, notable differences were observable. Most systematic reviews underscored the importance of genetic testing, which is considered as the main diagnostic tool in breast cancer. However, case series studies were more reliant on enzyme activity data. Interestingly, enzyme activity analysis was central in both types of studies; this implied that the enzyme activity was important in GD diagnoses. Upon this, we discovered patterns in the GD patient categories under study. Many systematic reviews concerned type-1 and type-3 GD patients, giving them special highlight in this regard. On the other hand, almost all the cases series were mainly Type-I GD patients although Type-II was also evident in both kinds of the studies. For instance, there was lack of type-3 GD patients in case series, which may result from different research attention. In looking at the marital status history of both patients’ parents, it was observed that similarities existed. Systematic reviews as well as case series studies demonstrated that the prevalence of consanguineous marriages was higher among parents of GD patients. Such commonality points toward genetic factor in GD prevalence. In conclusion, The comparison shows that despite the similarity of systematic reviews and case series studies in unraveling Gaucher’s Disease, there are minute differences. Such differences only serve to emphasize that there is a dire need for an overall perspective that takes into account not just the general overarching evidence but also the findings contained in individual case studies, thereby presenting the full picture of this complicated illness. The study aimed to investigate the effectiveness of patient counseling for individuals with Gaucher Disease in Tamilnadu, focusing on three crucial aspects: genetic factors, dietary modifications, and lifestyle changes. Challenges in dietary modifications include cultural dietary preferences, financial constraints, or lack of awareness regarding specific dietary requirements. Lifestyle changes, such as exercise and stress management, can significantly impact the overall well-being of patients with Gaucher Disease. The study highlights the importance of patient education and support in enhancing adherence to counseling recommendations. We obtain the score from the patient for before and after counselled by us and we received the outcomes like both positive and negative results. Then we created a Whatsapp group named as Gaucher Care Community forfollow up of patient counselling and keep interacting with the patients condition and quality of life and to spread positivity to the GD patients and also we included the picture of our whatsapp group below. Our study was compared to another study observed that One hundred twenty genetic counselors completed the quantitative survey, distributed in Fall of 2017. While the majority of respondents knew of the Gaucher-related Parkinson’s link (n?=?78; 65%), just over one-third reported discussing it in preconception/prenatal settings (n?=?30; 38.5%). Respondents reported discussing these links more consistently when disclosing positive results or when the patient/family approached the topic. Respondents cited the lack of professional guidelines as one of the main reasons for not discussing the link [110].
CONCLUSION
Systematic review:
The study has revealed the pattern of presentations observed in Gaucher Disease patients of Tamil Nadu. This study is instructive on the early symptoms of this rare hereditary disorder and adds to understanding of its clinical manifestation. Prenatal screening assessment makes it evident that one needs to do early detection and intervention. Early detection can prevent or delay symptoms, improve disease management, and reduce complications. This paper provides a thorough overview of the current evidence to enable development of targeted counseling guidelines for patients suffering from Gaucher Disease and their families in Tamil Nadu.
Case Series Study:
The case series study enabled us to take a closer look at the specific experiences of Gaucher Disease patients in Tamil Nadu. It sheds light on the challenges faced by these individuals and their efforts to manage the disease and improve their quality of life. Through case studies we learnt about the disease’s presentation in a particular area. This information will facilitate the development of special guidelines in counseling that will cater for the local specific needs facing the population.
Patient Counseling:
Further research is needed to validate and refine these prediction models and diagnostic strategies.our effective patient counseling is crucial for individuals with Gaucher Disease to manage their condition effectively. While genetic counseling can aid in understanding the disease's hereditary nature, dietary and lifestyle modifications are equally important for symptom management and overall well-being. The study highlights the need for tailored counseling guidance. Through these guidelines, the Gaucher Disease patients and their families are provided with the necessary information and guidance for managing the disease successfully
LIMITATIONS
Certainly, here are two key limitations of the case series study on Gaucher disease patients in Tamil Nadu:
ACKNOWLEGMENT:
We would sincerely thanks to DR. Sujatha Ph.D., (Women’s Empowerment); Mr.P.Naresh B.Sc (Advanced Zoology and Biotechnology) from MediScan, Mylapore, Chennai, Tamilnadu for assisting us to collect the cases.
REFERENCES

Anandhasayanam A. , Hanish Fathima S. , Sabarieshwaran K. , Jerin Mathew M. , A Case Series Study To Identify Early Prediction Possibilities & Develop Customized Counselling Guidelines For Tamil Nadu Gauchers Disease Patients/Family With A Systematic Review Of Evidence, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 7, 1226-1260. https://doi.org/10.5281/zenodo.12760909
 10.5281/zenodo.12760909
10.5281/zenodo.12760909