Lokmanya Tilak Institute of Pharmacy, Kharghar, Navi Mumbai, Maharashtra, India.
Lichen planus (LP) is a chronic inflammatory disease caused by the immune system, affecting the skin, mucous membranes, nails, and scalp. Histologically, it is marked by a T-cell-mediated assault on basal keratinocytes. The precise etiology remains unidentified; however, potential triggers encompass genetic susceptibility, viral infections such as Hepatitis C, diverse pharmacological agents, and stress. Clinically, it manifests variably according to the location. The "6 P's" are often used to describe cutaneous LP: pruritic, purple, polygonal, planar papules and plaques that often show up on flexor surfaces. Because oral lichen planus (OLP) has a transformation rate of roughly 0.4–3% and is regarded as a potentially malignant oral condition, it is vital. Other types include nail LP, which can result in permanent damage, and lichen planopilaris (scalp), which causes scarring alopecia. A skin biopsy demonstrating a distinctive lichenoid interface dermatitis confirms the diagnosis, which is mainly clinical and validated by dermoscopy. Topical and systemic corticosteroids, retinoids, calcineurin inhibitors, and non-pharmacological treatments like laser and phototherapy are all part of the management strategy, which tries to control symptoms.
Lichen planus (LP) is a chronic, immune-mediated inflammatory disorder affecting skin, mucous membranes, nails, scalp, and genital mucosa.[1] It is characterized histologically as a lichenoid (interface) dermatitis in which autoreactive T cells, especially CD8? cytotoxic T lymphocytes, target basal keratinocytes, leading to vacuolar degeneration of the basal layer and apoptosis (Civatte bodies). [2] Although the exact etiology is not fully clear, multiple factors are implicated — including genetic predisposition, autoimmune dysregulation, viral triggers (e.g. hepatitis C), drug exposures (e.g. antimalarials, β-blockers, thiazides, nonsteroidal antiinflammatory drugs), contact allergens, and psychological stress. [3] Notably, a meta-analysis and epidemiologic studies have shown a significant association between hepatitis C virus (HCV) infection and lichen planus, particularly oral LP, though the strength of association varies by region and study design. [3] The prevalence of cutaneous LP in the general population is variably estimated (often \~0.2–1%) depending on geographic region, while mucosal (oral) lichen planus may have a higher prevalence in some series. [4] The typical age of onset is between 30 and 60 years, although cases in younger and older individuals are reported. [3] There is often a slight female predominance in many series.[4] Oral lichen planus (OLP) is of particular clinical importance because it is classified by the World Health Organization among oral potentially malignant disorders. [5] The reported malignant transformation rate is low (commonly quoted around 0.4–3 %), but variable across studies.[4] Because of this, careful long-term surveillance is often recommended for patients with OLP. [5] LP is fundamentally a relapsing–remitting disease: many patients experience periods of exacerbation and remission over months to years. [4] Compared to cutaneous LP, mucosal disease (especially oral) tends to be more persistent, more resistant to therapy, and with a greater risk of chronicity. [5] Cutaneous lichen planus (CLP) represents the classic manifestation of the disease and is most often described using the mnemonic of the “6 P’s” — pruritic, purple (violaceous), polygonal, planar (flat-topped) papules and plaques.[3] Lesions are typically shiny, firm, and may vary in size from a few millimeters to over one centimeter. A hallmark diagnostic feature is the presence of Wickham’s striae, seen as delicate, white, lace-like streaks across the surface, which are accentuated under magnification or after topical alcohol application. [3], [6] The distribution of CLP tends to be bilateral and symmetrical, favoring the flexural surfaces of the wrists and forearms, the lower legs and ankles, and the lumbar region, while the trunk is less commonly affected. [3] The Koebner phenomenon is frequently observed, wherein new lesions appear along lines of trauma, scratches, or pressure sites. [3] The natural course of CLP is usually self-limited, with many cases undergoing spontaneous resolution within 1–2 years. However, recurrence is not uncommon, and residual post-inflammatory hyperpigmentation often persists, particularly in individuals with darker skin tones. [3], [4] Hypertrophic variants, in contrast, may remain for years and require long-term management. In sum, LP is a heterogeneous disease with multiple clinical phenotypes but unified by common immunologic and histopathologic features. In your article’s Discussion / Recent Advances section, you may wish to include newer data (for example, IL-17, JAK-STAT pathway, or biologic therapies) from recent reviews. [2]
Clinical Features:
The clinical manifestations of LP are diverse, depending on the tissue(s) involved. However, characteristic features are seen in the cutaneous and mucosal forms; additional involvement of nails, scalp, and genital areas is common in many patients.
Below is an expanded breakdown of clinical features site by site, with variant patterns:
“6 P’s” mnemonic: LP lesions are often described as pruritic, purple (violaceous), polygonal, planar (flat-topped), papules, plaques. [3] Papules/plaques are typically firm, shiny, and may measure from a few millimeters up to >1 cm. [6] A key diagnostic clue is Wickham’s striae — fine, white, reticular (lace-like) streaks seen on the surface of lesions, more visible under magnification or after whitening with alcohol. [3] Lesions often occur on flexor surfaces (wrists, forearms), lower legs (shins, ankles), lumbar region, and less commonly trunk. [3] The Koebner (isomorphic) phenomenon is often present: new lesions can arise in sites of trauma, scratches, or pressure. [3] Lesions may be arranged linearly, in annular configurations, as hypertrophic (verrucous) plaques especially on shins, or less commonly in atrophic form.[3] Hypertrophic LP is notably more pruritic and persistent; residual pigmentary changes (post-inflammatory hyperpigmentation) are frequent after resolution. [4] Many cutaneous lesions may resolve spontaneously over 1–2 years, though recurrence is common, and pigmentary sequelae (hyperpigmented macules) often persist. [3] Rare forms include actinic LP (sun-exposed distribution), pigmented LP, linear LP (along Blaschko’s lines), and bullous LP. [6]
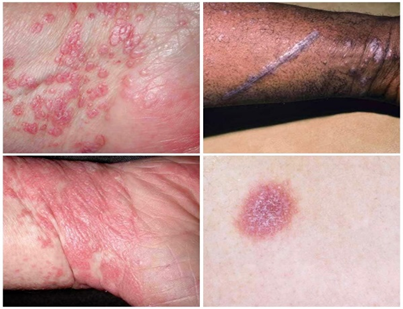
Fig.no:1 Cutaneous lichen planus [8]
Oral involvement occurs in a substantial subset (various studies report \~50?70 % or more) and may precede, coexist, or occur in isolation from skin disease. [7] The most common morphological form is reticular (reticulated) LP, characterized by bilateral interlacing white striae (Wickham’s striae) on buccal mucosa, gingiva, tongue, lips, and less often palate. [7] Other forms include atrophic, erosive, ulcerative, plaque, and rarely bullous. The erosive variant is symptomatic (pain, burning), often persistent, and associated with higher malignant risk. [5] Symptoms may include burning sensation, pain with hot/spicy foods, sensitivity, dysgeusia, odynophagia (if lesions extensive), or desquamative gingivitis (especially on gums). [5]The natural course is usually chronic; some lesions wax and wane, but full remissions are less frequent than in cutaneous disease. [5] Malignant transformation (typically to oral squamous cell carcinoma) is reported in some longitudinal studies; reported rates vary (commonly \~0.4–3 %). [4] It is thus recommended to monitor erosive and atrophic cases, especially those with risk factors (smoking, HPV, dysplasia). [7]
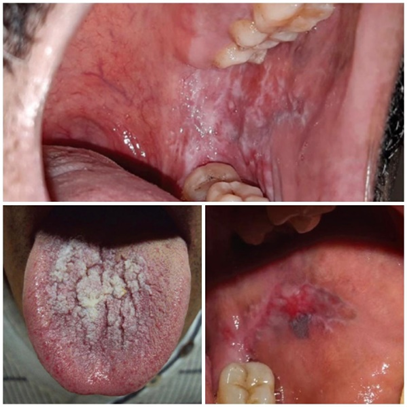
Fig.no:2 Oral lichen planus [5]
Nail involvement is less common but clinically significant (often cited in \~5–10 % of patients). [3] Clinical features include longitudinal ridging/grooving, thinning, onychorrhexis, subungual hyperkeratosis, pterygium formation (scarring of proximal nail fold onto matrix), onycholysis, nail shedding, or even anonychia (complete loss). [6] Early recognition is important, as advanced changes may be irreversible. [4] Sometimes only the nail is affected (without obvious skin or mucosal disease). [1]
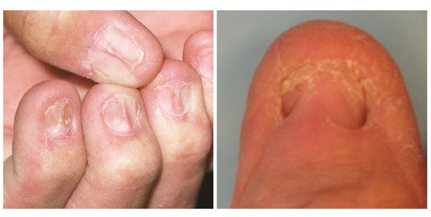
Fig no :3 Nail lichen planus[4]
When LP affects the scalp, it is commonly referred to as lichen planopilaris (LPP). [4] Clinical features include perifollicular erythema, scaling, follicular hyperkeratosis, and often cicatricial (scarring) alopecia. [6] Over time, the destruction of hair follicles leads to permanent hair loss in affected regions (patchy or confluent). [4] A variant, frontal fibrosing alopecia, is sometimes considered part of the LPP spectrum. [6]
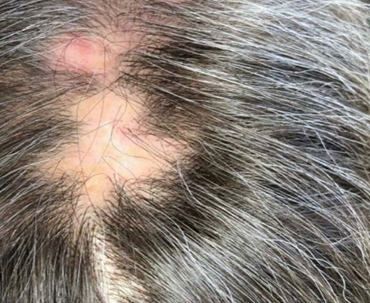
Fig.no:4 Scalp (Lichen Planopilaris)[8]
LP can involve male and female genital mucosa. In women, vulvovaginal LP may present as erosive or papular lesions, causing pain, burning, dyspareunia, and may lead to scarring, fusion, introital stenosis. [3] In men, classic penile LP may present as annular papules on the glans, or occasionally erosive or whitish streaks. [3] Overlap with lichen sclerosus or other chronic vulvovaginal conditions is not uncommon, complicating diagnosis. [4] The risk of malignant transformation is also considered in genital LP, albeit lower in frequency. [4]
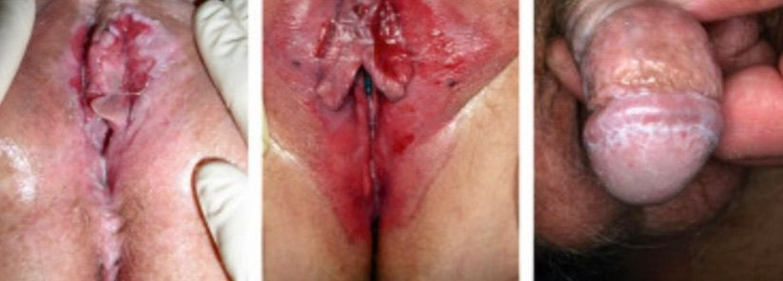
Fig.no:5 Genital lichen planus [1]
Actinic (photosensitive) LP: lesions in sun-exposed areas (face, dorsal hands), sometimes with pigmentary changes; usually non-itchy, and without mucosal involvement. [6] Pigmented LP / Lichen planus pigmentosus: characterized largely by hyperpigmented macules (brown, grey) in sun-protected or sun-exposed zones, often without a preceding active inflammatory stage. [8] Linear LP: lesions following Blaschko’s lines. [6] Annular/atrophic/vesiculobullous / hypertrophic (verrucous) forms as noted above. [6] Lichenoid drug eruptions / lichenoid reactions: drug-induced LP-like eruptions should always be in the differential, often with subtle clinical differences and sometimes a photo-distributed pattern.[6] Lichen planus pemphigoides: rare overlap with bullous pemphigoid, with blister formation both at LP sites and distant skin, due to epitope spreading. [6]
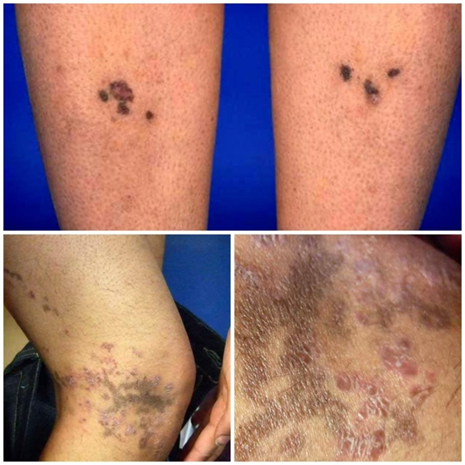
Fig.no 6 Other/Less common variants of lichen planus [8]
Natural History, Course & Prognosis:
Cutaneous LP lesions often resolve spontaneously within 1–2 years in many patients, especially in non-mucosal disease. However, recurrences are common.[3] Mucosal (especially oral) LP tends to be more persistent and recalcitrant; complete remission is less common[5]. Post-inflammatory hyperpigmentation frequently persists long after the active lesions resolve. [4] In scalp (LPP), hair loss is often permanent once follicular destruction occurs. [6] In erosive mucosal or genital LP, chronicity, pain, scarring, and malignant transformation risk (though low) are ongoing concerns. [4]
EPIDEMIOLOGY:
There are between 0.2% and 1% of adults worldwide who have cutaneous lichen planus. An estimated 1% to 4% of people have oral lichen planus, which is more prevalent. At a ratio of 1.5:1, women are generally more afflicted than men, and the majority of cases occur between the ages of 30 and 60. Less than 5% of all afflicted individuals are youngsters, making the illness extremely uncommon. [9] Despite the fact that lichen planus is not thought to have an ethnic preference, new research indicates that African Americans and people of Indian and Arabian heritage may be more susceptible to the disease. Given that up to 10% of patients' first-degree relatives may also have the illness, there seems to be a familial component. [9] A recent meta-analysis of 46 studies found that the prevalence of LP is 0.89% in the general population and 0.98% in patients seeking dermatological care. In the majority of study groups, oral LP outnumbers cutaneous LP, which has been shown to affect 0.2% to 1.0% of the adult population. With a range of 14 to 250 cases/100,000 person-years, the incidence of LP is less well defined and exhibits significant geographic variation (25–29). Rather than reflecting the presence of an ethnic predisposition, this variability most likely reflects methodological variations in the sampled groups. [1] Women are more likely than men to experience oral LP. [1]
Lichen planus may present in patients as varied cutaneous manifestations alone or in association with mucosal lichen planus and, or lichen planus of nails. Research indicates frequency of involvement of mucosa in lichen planus patients to be 30- 70%. Oral lichen planus is quite prevalent, It is amongst the most prevalent mucosal disease. The incidence in the general population is approximately 1.27–2.0%, with an estimated 6-7 million individuals influenced in the United States alone. [11] The disease is found mostly in middle-aged people (30–60 years) with a female-to-male distribution of 2:1.[11] Oral lichen planus in children is uncommon. Approximately 50% of females with oral lichen planus were found to have undiagnosed vulvar lichen planus.[11]
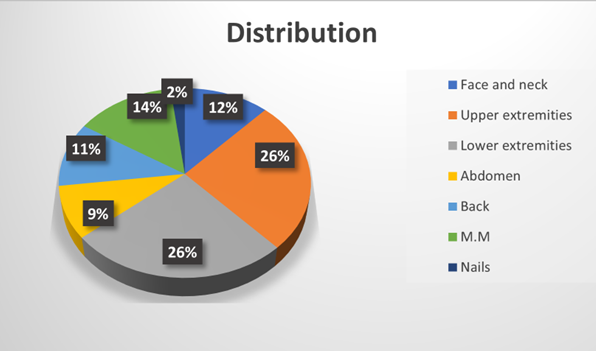
Fig.no 7 Anatomical distribution of lichen planus[13]
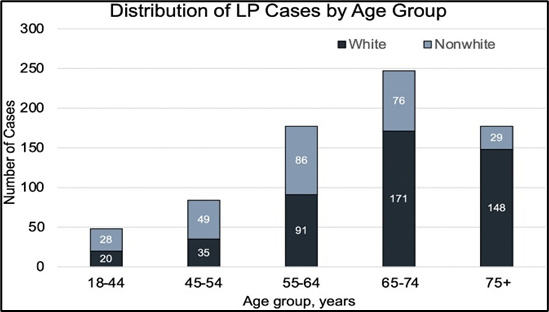
Fig.no 8 Distribution of LP cases by age group [10]
About 3.6 million persons were analyzed in 2015 and 2018. Within this population, a prevalence of LP of 90.3 LP cases per 100,000 persons in 2015 and of 104.4 cases per 100,000 persons in 2018 was found within the database. The incidence rate of LP – adjusted for the entire German population – was 84.7 and 95.9 per 100,000 persons in 2015 and 2018, respectively. This aligned with a combined estimated figure of 69,591 LP cases in Germany during 2015 and 79,605 LP cases during 2018 [12] The total prevalence of hypertrophic LP, lichen planus pemphigoides and lichen planopilaris (LPP) extrapolated to the German population was 3.0 per 100,000, 0.4 per 100,000 and 5.2 per 100,000, respectively, constituting 2.81 %, 0.40 % and 5.04 % of all prevalent LP patients in 2018. The total incidence of hypertrophic LP, lichen planus pemphigoides and LPP was 0.6 per 100,000, < 0.1 per 100,000 and 1.6 per 100,000 individuals in 2018, respectively. A majority of the diagnoses were recorded as LP without additional specification (L43.9 LP, unspecified: > 60 %). Out of all the prevalent LP patients in 2018, 9.99 % had more than one subtype of LP diagnosed. The total prevalence of LDR was 0.6 per 100,000 and the total incidence of LDR was 0.6 per 100,000 individuals.[12] They reported a total age-adjusted prevalence of 1.27% (0.96% in males and 1.57% in females) in Sweden. LP incidence was 0.032%–0.037% in one British population. LP generally occurs in middle-aged patients of both sexes. There is no sexual predilection but some series show a slight female predominance up to the ratio of 2 : 1. Interestingly, in the 3 largest case series of childhood LP, there is reported to be a female to male ratio of 1 : 2 in a US population ,1 : 1.5 in an Indian cohort, and 2 : 1 in a Canadian study. [4] The reasons for such inconsistencies may lie in varying inclusion and exclusion criteria in the studies. The relative male predilection in childhood LP is not typical for an autoimmune disorder and implies that there are possibly other yet unknown mechanisms involved in LP pathogenesis. Childhood LP occurs more frequently in the African American population. Additionally, hypertrophic and actinic types as well as LP pigmentosus occur more often among African Americans or dark-skinned people. [4]
ETIOLOGY:
Even though the exact cause of lichen planus is unknown, it seems to be an idiopathic disease mediated by T cells. According to the widely accepted theory, cytotoxic T cells are activated when epidermal self-antigens are changed by exposure to an exogenous agent, such as a virus, medication, or contact allergen. T-cell targeting and apoptosis are the outcomes of the altered self-antigens' cross-reaction with basal keratinocytes' normal self-antigens. Lichen planus has been associated with several agents, with a particular focus on its relationship with viruses, notably the hepatitis C virus (HCV). HCV seropositivity increases the risk of developing lichen planus by 2.5 to 4.5 times, and patients with lichen planus are five times more likely to test positive for HCV than the general population.[14] Lichen planus and the COVID-19 virus and vaccine have recently been connected.[15] Contact allergies to several metals present in dental restorations, such as copper, gold, and mercury, are linked to oral lichen planus. There have been reports of lichen planus lesions clearing up after the sensitizing metal is removed.[17] Although many medications are linked to lichen planus, it is uncommon for lesions to get worse after repeated exposure to the medication. Angiotensin-converting enzyme inhibitors, thiazide diuretics, quinidine, beta-blockers, tumor necrosis factor-α inhibitors, antimalarials, and gold are more frequently linked medications.[17]
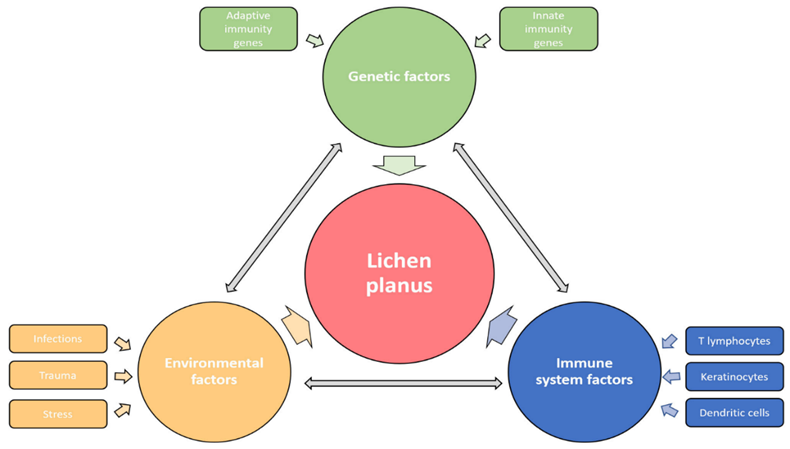
Fig.no 9 Etiological factors [16]
PATHOGENSIS:
Since there are typically no studies on this disease in humans or animals, the etiopathogenesis of cutaneous LP, a rare dermatosis, remains unclear[18]. According to current knowledge, LP immunopathogenesis is caused by the immune or autoimmune reaction of T cells to exogenous or self-altered antigens that are presented by antigen-presenting cells (APCs), such as keratinocytes or dendritic cells (DCs) [18]. Following the release of damage-associated molecular patterns (DAMPs), like S100A8/A9, which activate Toll-like receptors (TLRs), the initiation phase starts. This process causes plasmacytoid DCs (pDCs) to secrete a large amount of type I interferon (IFN-α), which is further supported by keratinocyte-produced IL-1β and TNF-α [19]. IFN -α stimulates inflammatory dermal DCs (dDCs), which migrate to the surrounding lymph nodes, and upregulates chemokines like CXCL9 and CXCL10. There, they expose naïve T lymphocytes to the antigens and, through the release of interleukin (IL)-12 or IL-23, encourage the development and growth of T1 helper and cytotoxic (Th1 and Tc1) and T17 (Th17 and Tc17) lymphocyte subpopulations [19]. In the early stages of the disease, keratinocytes, APCs, and T lymphocytes interact to produce more Th1 cytokines, like IFN-γ, which is a crucial event in LP [18].
Attracted by cytokines and chemokines derived from innate cells and aided by increased expression of adhesion molecules like LFA-1, ICAM-1, and VCAM-1 as well as basement membrane extracellular matrix proteins, T1 and T17 lymphocytes, which express skin-homing receptors, exit the circulation and migrate to the site of inflammation during the progression phase [18]. When memory T cells are stimulated, they produce a lot of cytokines, such as Th1's IFN-γ, TNF-α, and IL-2, or IL-17 and IL-22. In conjunction with TGF-β and IL-6, IL-17 promotes further Th17 expansion [20]. Thus, this intricate cascade reinforces the vicious cycle of inflammation by supporting pathways linked to Th1 and Th17 [20].The generation of oxygen free radicals (ROS), which trigger cell death, is the end result of pro-inflammatory cytokines. The role of oxidative stress has been hypothesized in light of the prooxidant–antioxidant imbalance observed in LP. But as was already mentioned, Tc action plays a crucial role in the pathophysiology of LP [21]. Specifically, Tc1 and Tc17 effector cell activation triggers the caspase cascade through the interaction of TNF-α and TNF-α-R1, Fas and FasL, and the release of cytotoxic molecules from the cytoplasmic granules, including granulysin, granzyme B, and perforin [18]. These strong lytic particles mediate Tc-induced epidermal damage, which results in the apoptosis of basal keratinocytes and the emergence of the distinctive LP phenotype [18]. Granule exocytosis is a major pathway of cytotoxicity in humans, according to conducted studies, although other mechanisms complement this mechanism at different stages of the disease. While recent studies have confirmed granulysin overexpression in the skin and peripheral blood of LP patients, previous studies have found elevated levels of TNF-α in sera and Fas-FasL, granzyme B, and perforin in the skin compared to healthy controls. Patients with psoriasis have shown a similar finding, indicating that the pathogenetic mechanisms of these chronic inflammatory dermatoses are similar[16]. In LP, inflammatory cell death is decreased, which aids in the advancement of the inflammatory process, while epithelial cell apoptosis is highlighted [18]. Prior findings demonstrated the significance of the crosstalk between CD8+ and CD4+ T cells, as their interaction is necessary for the activation of cytotoxicity in the afflicted skin [18]. CD8+ T lymphocytes express the molecule of request for cytotoxic activity (RCA) and release mediators to draw in additional inflammatory cells after identifying antigens expressed by keratinocytes via MHC-I molecules [16]. In the meantime, CD4+ T cells, which are activated by DC contact and IL-12, express RCA receptors and release IL-2 and IFN-γ, which indirectly activates and multiplies CD8+ T cells [16].
The condition is further complicated by tissue-resident memory T cells (TRM) in addition to effector cells that arrive by blood. Because TRM can be repeatedly activated at the site of previous inflammation, resulting in LP reactivation and flare, they create a local skin reservoir of chronic inflammation [22]. The aforementioned mediators also draw regulatory T cells (Tregs) to the skin. By interfering with effector T lymphocytes' interactions with DCs, they typically play a significant part in immunosurveillance and effector T lymphocyte suppression. But because Tregs are flawed in LP, they are unable to effectively manage the illness [23]. T lymphocytes have close interactions with other immunocytes, including mast cells, NK cells, and macrophages, which also take part in this intricate inflammatory cascade [19].The action of mast cells, chemokines and matrix metalloproteinases (MMPs) contributes to nonspecific immune mechanisms. [16] In the lesional dermis, especially the basement membrane rupture area, there is an increased amount of degranulated mast cells releasing numerous mediators, such as IL-3, IL-4, IL-5, IL-6, IL-8, IL-10, IL-16, TNF-α and chymase, and RANTES [24]. RANTES (CCL5) is part of the chemokine network, which is, in addition to mast cells, produced by T lymphocytes and keratinocytes to stimulate the inflammatory cells’ migration. The importance of the CCL5-CCR5 axis has been recently pointed out by Shan et al., who reported its role in establishing LP chronicity. [16] Chemokines CXCL-9 and CXCL-10, chemokine receptors CXCR3 and CCR5, and members of the endoprotease family, MMP-2, MMP-7 and MMP-9, are also present in increased concentrations in LP [16]. Protease chymase stimulates lymphocytes’ production of MMP-9, whereby mast cells indirectly contribute to basement membrane disruption [24].
HISTOPATHOLOGY:
LP is characterized by lichenoid interface dermatitis. The basic histological findings include a thick, continuous, and band-like lymphohistiocytic infiltration at the dermal-epidermal junction and in the upper dermis. The infiltrate conceals the dermal-epidermal junction and makes it difficult to identify the basal layer in the early stages of the disease. In LP lesions, epidermal alterations include irregular epidermal hyperplasia with a jagged "sawtooth" look, compact hyperkeratosis or orthokeratosis, foci of wedge-shaped hypergranulosis, basilar vacuolar degeneration, minor spongiosis in the spinous layer, and squamatization. The dermal papillae between the elongated rete ridges are commonly dome-shaped. Necrotic keratinocytes are seen at the dermal-epidermal junction and in the basal layer of the epidermis. "Colloid or civatte bodies" are eosinophilic remains of anucleate apoptotic basal cells that can also be seen in the dermis. The regions of hypergranulosis are typically where Wickham striae are observed. There may be focal subepidermal clefts (Max Joseph spaces) as a result of vacuolar degeneration at the basal layer. Cells in the basal layer mature and flatten, which leads to squamatization. It occurs where the sawtooth pattern of rete ridges is prominent and there is noticeable hypergranulosis. Hair follicles (acrotrichia) or eccrine ducts can both develop wedge-shaped hypergranulosis. The related hyperkeratosis, parakeratosis, hypergranulosis, papillomatosis, acanthosis, and hyperplasia significantly increased in the hypertrophic subtype, as thicker collagen bundles formed in the dermis. Moreover, in contrast to the usual sawtooth pattern, the rete ridges are more expanded and rounder. Retinal ridge loss and skin fibrosis are common in atrophic LP. The course of vesiculobullous LP is faster. Because of this, certain characteristics, such as hyperkeratosis, hypergranulosis, or dense lymphocytic dermal-epidermal infiltration, might not be included. If the number of melanophages in the papillary dermis continues to rise, LP lesions may heal with residual hyperpigmentation. [4]
The lower section of the hair follicle is spared in classic Lichen planus pigmentosus (LPP), as the bandlike lymphocytic infiltrate is originally restricted in the peribulge area, including the infundibulum and isthmus. Orthokeratosis, hypergranulosis, and follicular plugging can all occur in the same follicular segments. Seldom is the interfollicular epidermis impacted. Because hair follicle stem cells are involved in the bulge, LPP causes irreversible hair loss. Hair follicles may gradually be replaced by fibrotic tissue. [4] ,[25] The epithelial alterations in mucosal lesions are less distinct. Because normal oral mucosa has parakeratosis without a granular layer, the rete ridges lack the distinctively prominent sawtooth pattern. Thus, orthokeratosis is uncommon in OLP lesions. Compared to CLP, OLP lesions are probably more atrophic than acanthotic. Histopathological findings in genital erosive LP are less specific and frequently inconclusive than those in CLP and OLP [4]
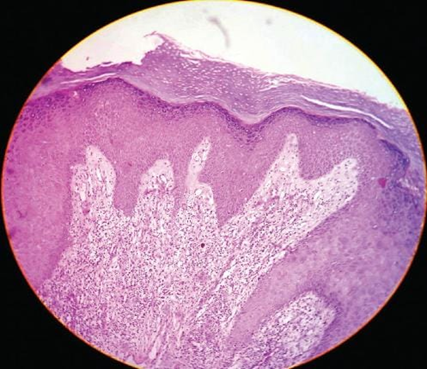
Fig.no 10 Wedge-shaped hypergranulosis[26]
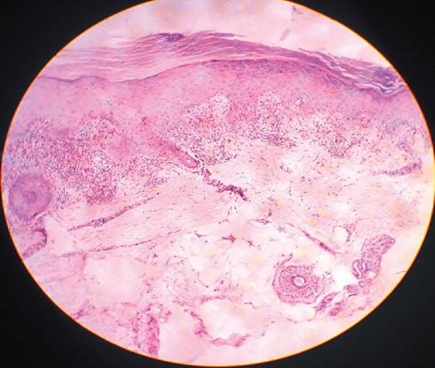
Fig.no 11 Dense dermal infiltrate[26]
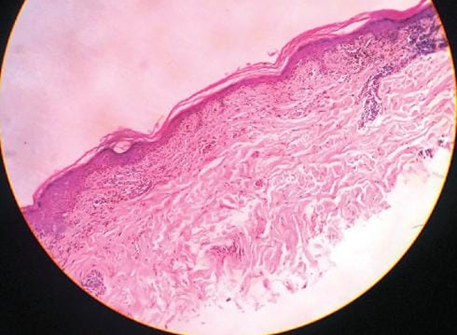
Fig.no 12 Squamatisation of basal layer with atrophic epidermis[26]
INVESTIGATIONS:
MANAGEMENT OF LICHEN PLANUS:
Pharmacological Treatment:
1. Corticosteroids -
2. Calcineurin Inhibitors -
3. Retinoids -
4. Dapsone -
5. Mycophenolates -
6. Low-Dose Heparin (Enoxaparin) -
7. Biologics (Efalizumab) -
Non-Pharmacological Treatment:
1. PUVA Therapy (Psoralen + UVA) -
2. Photodynamic Therapy (PDT) -
3. Laser Therapy -
CONCLUSION:
A complicated and multifaceted inflammatory illness, lichen planus is distinguished by its unique immunologic and histologic characteristics. Its clinical manifestation is quite diverse, affecting various body parts in various ways, ranging from the traditional cutaneous papules to the more persistent and worrisome erosive mouth lesions. Although recurrences are frequent, the disease has a relapsing-remitting course, with cutaneous forms frequently going away on their own in 1-2 years. OLP in particular is a mucosal disease that is more likely to be chronic and treatment-resistant. Long-term monitoring for possible malignant development into squamous cell carcinoma is a crucial component in managing LP, especially the oral variety. A variety of topical, systemic, and medical therapies are used to treat the disease, with an emphasis on symptom relief and inflammation control, depending on its severity and location. The main focus of both established and new therapies continues to be the underlying T-cell-mediated pathophysiology.
REFERENCES

N. Bubera, S. Khandare, T. Palte, K. Sirvi, S. Jadhav, A. Khochare, S. Patil, An Overview of Lichen Planus: From Etiological Factors to Therapeutic Strategies, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 10, 1892-1907. https://doi.org/10.5281/zenodo.17379574
 10.5281/zenodo.17379574
10.5281/zenodo.17379574
