
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Dr. Gurachar Nargund College of pharmacy Muradi Koppal.
Tuberculosis, caused by Mycobacterium tuberculosis, remains a critical global health concern, particularly in the face of increasing resistance to first-line drugs. In this study, a series of novel coumarin derivatives (3a to 3e) was designed and evaluated for their inhibitory potential against the enoyl-acyl carrier protein reductase (InhA), a validated drug target involved in mycolic acid biosynthesis in M. tuberculosis. Molecular docking studies were conducted using Autodocking to assess binding affinity and interaction profiles of the compounds within the active site of InhA. Among the synthesized derivatives, compound 3d exhibited the highest binding affinity with a docking score of -5.91 kcal/mol, significantly better than the standard drug isoniazid (INH), which showed a docking score of -4.71 kcal/mol. The binding mode of 3d revealed key interactions with catalytic residues, supporting its potential as an InhA inhibitor. Furthermore, ADME predictions indicated that all tested compounds, including 3d, possess favorable pharmacokinetic and toxicity profiles. These findings suggest that coumarin-based scaffolds, particularly compound 3d, hold promise for further development as novel anti-tubercular agents targeting InhA.
Tuberculosis
Tuberculosis (TB) is an acute or chronic infectiousdisease caused by several Mycobacterium species, collectively called tubercle bacilli or Mycobacterium tuberculosis complex, which includes Mycobacterium tuberculosis [MTB] itself, Mycobacterium microti, Mycobacterium pinnipedii, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium africanum and Mycobacterium canetti. MTB is also known as the whiteplague. Robert Koch first identified in 1882. TB affects thelungs, bones, joints, and even the skin. It affects the central nervous system, lymphatic, circulatory, genitourinary andgastrointestinal systems too. Tuberculosis (TB) is acommunicable disease that is a major cause of ill healthworld wide and the leading cause of death from a singleinfectious agent [1]. An estimated 10.8 million people fell ill with TB in 2023, with 134 Incident cases per 100,000 population. TB deaths is from 2021, with an estimated 1.6 million deaths, including 1.4 million among HIV-negative people and 187,000 among HIV-positive people[2]. Signs and symptoms, includes cough, haemoptysis, dyspnoea, chest pain, night sweating, anaemia, tachycardia, lung-auscultation finding, fever, low body-mass index, low mid-upper arm circumference giving patients a TB score from 0 to 13.[3] The intrinsic resistance of mycobacteria against several classes of antibiotics has commonly been attributed to the unusual composition and structure of the mycobacterial cell envelope. Compared to other gram-positive bacteria, the cell wall of members of the genus Mycobacterium is much thicker and more hydrophobic, due to the presence of a wide array of different lipids that include mycolic acids. Many studies performed in different mycobacterial species demonstrated that the composition of the cell envelope and the low numbers of porinscontribute significantly to the cell envelope's low compound permeability. A major constituent of the cell wall is a layer of lipids, which are covalently linked to the peptidoglycan layer via arabinogalactan. Furthermore, the cell wall contains ‘extractable’ immunogenic glycolipids. The lipid-rich nature renders the cell wall extremely hydrophobic and prevents the permeation of hydrophilic compounds. It is thought that small hydrophilic compounds, including many antibiotics active against M.tuberculosis, can only traverse the cell wall via water-filled porins .[4]
Causes:
Symptoms:
Coumarin:
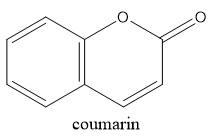
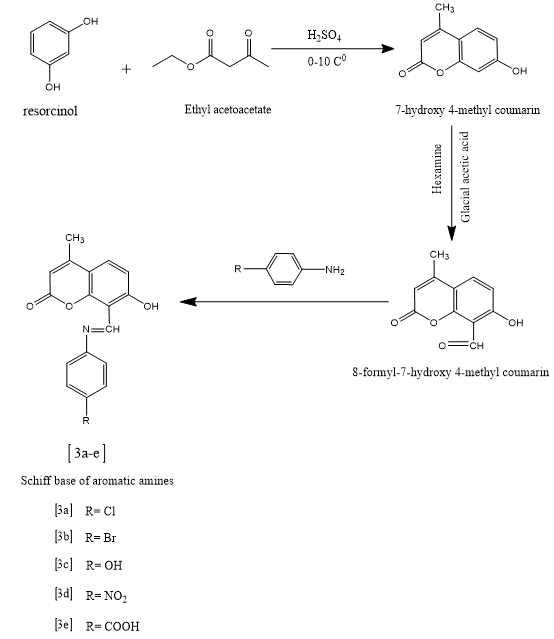
Coumarin is an oxygen heterocycle belonging to the biologically active class of compounds as it shows diverse biological activities . It naturally occurs as lactones and is also being used as perfumes and food flavouring agents. The numbering of coumarin starts from the ring oxygen, i.e. oxygen receives position-1, carbonyl carbon-2 and goes anticlock wise along with the ring. The parent coumarin was first isolated from Tonka beans by Vogel in the year 1820 . For the last ten years, the synthesis of coumarin derivatives has been the core of attention for medicinal chemists around the globe. Coumarin derivatives have great significant due to theirwidespread pharmaceutical activities. They have benefittedthe medical world by their outstanding performance as antidepressant,anti-HIV, antioxidant , antimicrobial , anti-inflammatory, antinociceptive,anti-influenz, antitumor, antiviral, antituberculosis, anti-Alzheimer, antihyperlipidemic , antipyretic, antiasthmatetc.Some of the coumarin derivatives, which are alreadysuccessful in the market are Brodifacoum (anticoagulant),Warfarin (anticoagulant), Armillarisin(antibiotic),Bergapten (sun screening agent), Auraptene (chemopreventativeagent), Acenocoumarol (anticoagulant),Ensaculin(KA672) (NMDA antagonist and a 5HT1A agonist), Hymecromone (choleretic and antispasmodic),Difenacoum (anticoagulant), Carbochromen (coronarydisease), Scopoletin, Phenprocoumon (anticoagulant).[1]
Aromatic amines:
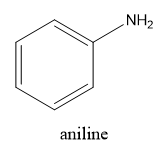
The aromatic amines are an industrially important class of organic compounds being widely used in the production of compounds and intermediates for the dyestuff, rubber-processing, and drug-manufacturing industries. Most aromatic amines possess the expected basic character that results from the presence of the lone pair of electrons on the nitrogen atom and like the aliphatic amines, they usually form stable salts with inorganic and also with many organic acids. They are, however, considerably weaker bases than ammonia or the aliphatic amines, and this reduced basicity may be interpreted as resulting from the direct attachment of the nitrogen atom to the aromatic system. Primary aromatic amines undergo N-alkylation on heating with an alkyl halide to give successively secondary amines, tertiary amines, and, finally, quaternary ammonium halides. Primary aromatic amines condense with aromatic aldehydes, but less readily with ketones, to form anils (Schiff bases). Tertiary amines cannot react with aldehydes to form anils.[5] Aniline is a aromatic compound with molecular formula C6H7N, appears as a yellowish to brownish oily liquid with a musty fishy odor and Burning taste. Its melting point -6 °C; boiling point 184 °C;.Denser than water (1.022 at 68 °F) ; slightly soluble in water and vapor Pressure 0.67 mmHg at 77 °F . Toxic by skin absorption and inhalation. [6]
Enoyl acyl carrier protein reductase (InhA) mechanism (PDB ID: 5W07)
Enoyl Acyl Carrier Protein (ACP) Reductase (InhA) is an enzyme involved in fatty acid synthesis mainly mycolic acid biosynthesis . It is a part of Tyrosine-dependent oxidoreductase also known as a short dehydrogenase/reductase family, especially to NADH-dependent enoyl ACP reductase. It catalyses trans double bond reduction which is linked to a carbonyl group of an intermediate that is covalently linked to an acyl carrier protein in the FAS-II pathway. InhA is unique and promising target for development of anti-TB drugs due to various reasons, it possesses a conserved active site which is not observed in several other targets in Mtb. The deeper binding pockets in the active site provides with vast scope of design and development of wide a variety of small molecule inhibitors to be accommodated. It is the main target of antitubercular drug, isoniazid and do not have human ortholog. It allows the bypass of resistance problems. In fatty acid synthesis, InhA not only complete reaction cycle but also give out to attract reversible reaction processing carbon atom C 3 of acyl chain. Therefore, targeting InhA remains as a target of choice for development of drug molecule against Mycobacterial tuberculosis.[7]

Computer Aided Drug Discovery
Drug discovery and developing a new medicine is a long, complex, costly and highly risky process that has few peers in the commercial world. This is why computer aided drug design (CADD) approaches are being widely used in the pharmaceutical industry to accelerate the process. The cost benefit of using computational tools in the lead optimization phase of drug development is substantial. On an average, it takes 10-15 years and US $500-800 million to introduce a drug into the market, with synthesis and testing of lead analogs being a large contributor to that sum. Therefore, it is beneficial to apply computational tools in hit to lead optimization to cover a wider chemical space while reducing the number of compounds that must be synthesized and tested in vitro. The computational optimization of a hit compound involves a structure-based analysis of docking poses and energy profiles for hit analogs, ligand-based screening for compounds with similar chemical structure or improved predicted biological activity, or prediction of favorable affinity or optimize drug metabolism and pharmacokinetics (DMPK) or absorption, distribution, metabolism, excretion, and the potential for toxicity (ADMET) properties. The comparably low cost of CADD compared with chemical synthesis and biological characterization of compounds make these methods attractive to focus, reduce, and diversify the chemical space that is explored.
Molecular Docking
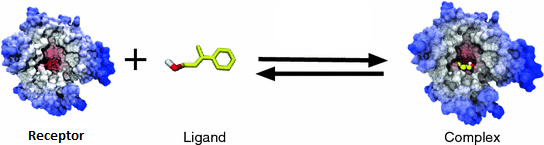
Molecular Docking is a method which anticipates the favored orientation of ligand (Drug candidate) against receptor (Protein) to make a stable complex. Favored orientation is possibly utilized to predict the strength of connection or binding affinity among ligand and protein by utilizing scoring functions. Docking is often applied to anticipate the binding orientation of drug candidates against protein targets in order to predict the affinity and activity of the drug. Therefore, docking plays a pivotal role in the drug design and discovery process. The main aim of molecular docking is to computationally simulate the molecular identification process and accomplish an optimized conformation so that the free energy of overall system is minimized. The process of discovery of a new drug is a very difficult task. Modern drug discovery is mainly based In-silico chemico biological approach. Use of computer aided techniques in drug discovery and development process is rapidly gaining popularity, implementation and appreciation.[9]
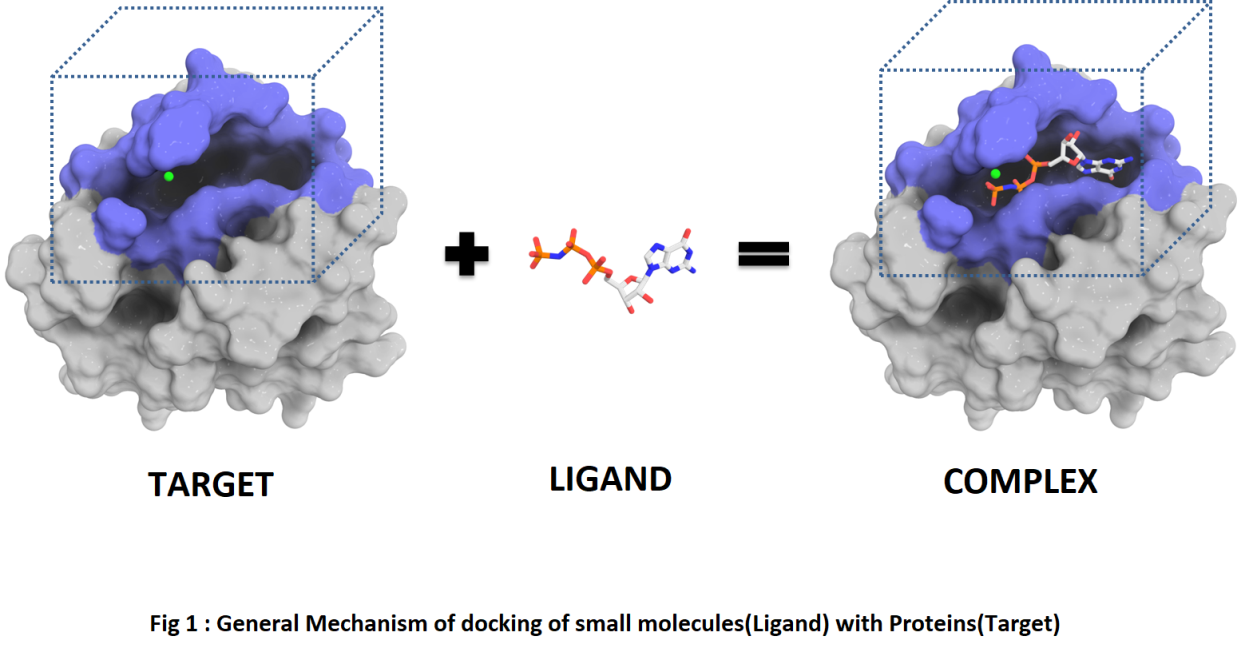
Molecular Modeling
Molecular modeling is a technique for deriving, representing and manipulating the structures and reactions of molecules, and those properties that are dependent on these three-dimensional structures in molecular modeling.[14]

Types of Docking [14]
There are 2 types of docking
1. Rigid Docking
If we assume that the molecules are rigid, then we are looking for a transformation in 3D space of one of the molecules which brings it to an optimal fit with the other molecules in terms of a scoring function. Conformation of the ligand may be generated in the absence of receptor or in the presence of receptor binding activity.[14]
We consider molecule flexibility then in addition to transformation, our aim to find the confirmations of the receptor and the ligand molecules, as they appear in complex in Figure 3[14]

Molecular Docking Models
The Lock and Key Theory
As far back as 1890 Emil Fischer proposed a model called the "lock-and-key model" as shown in Figure 4 states that explained how biological systems function. A substrate fits into the active site of a macromolecule, just like a key fit into a lock. Biological locks have unique stereo chemical features that are necessary to their function in following Figure 4.[15]

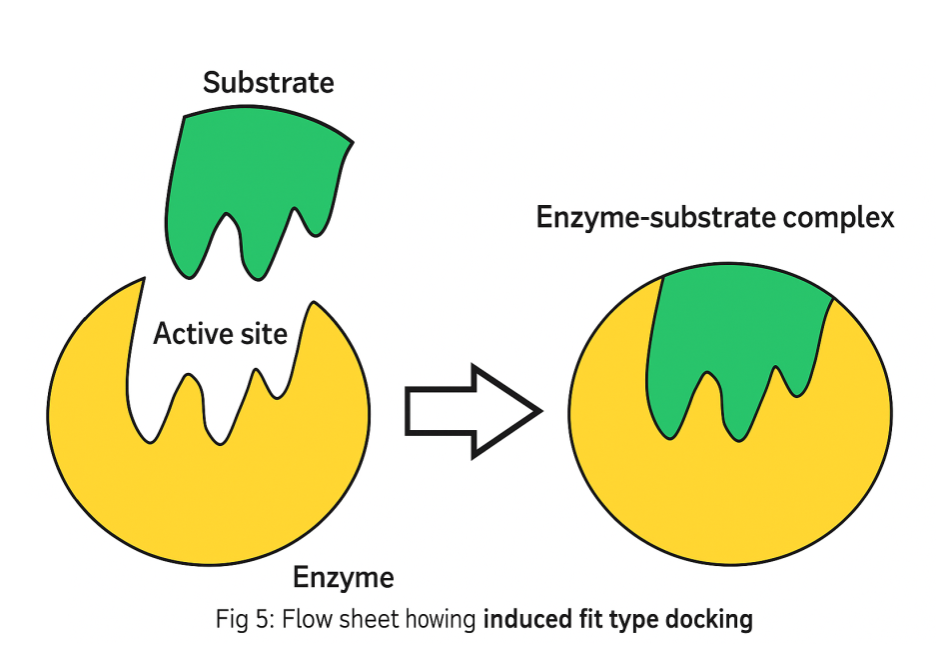
The Induced-Fit Theory
In 1958 Daniel Koshland introduced the "induced-fit theory". The basic idea is that in the recognition process, both ligand and target as shown in Figure 5 mutually adapt to each other through small conformational changes, until an optimal fit is achieved in following figure 5[15]
The Conformation Ensemble Model
In addition to small induced-fit adaptation, it has been observed that proteins can undergo much larger conformational changes. A recent model describes proteins as a pre-existing ensemble of conformational states. The plasticity of the protein allows it to switch from one state to another.[15]
Basic Requirements for Molecular Docking
The setup for a ligand docking approach requires components a target protein structure, the molecules of interest or a database containing existing o components a target protein structure, the molecules virtual compounds for the docking process, and a computational framework that allows the implementation of the desired docking and scoring procedures. Most docking algorithms assume protein to be rigid; the ligand is mostly regarded flexible. Beside the conformational degree of freedom, the binding pose in proteins binding pocket must be taken in to consideration. Docking can be performed by rigid molecules or fragments in to protein active site using different approaches like the clique-search, geometric hashing, pose clustering. Typically, the structure most likely to be dominant further adjusted by adding or removing hydrogens provided approximate pKa values. It is important to make sure that accurate atom typing occurs.[15]
Receptor Representation
The quality of receptor structure employed play’s central role in determining the success of docking calculations. In general, the higher the resolution of the employed crystal structure better will be the observed docking results. A recent review for accuracy, limitations and pitfalls of the structure refinement protocols of protein ligand complexes in general provided a critical assessment of the available structures.[15]
Mechanism of Docking
To perform a docking screen, the first requirement is a structure of the protein of interest. Usually, the structure has been determined using a biophysical technique such as X-ray crystallography, or less often, NMR spectroscopy. This protein structure and a database of ligands serve as inputs to a docking program. The success of a docking program depends on two components such as search algorithm and scoring function.[15]
Searching Conformational Space
The search space consists of all possible orientations and conformations of the protein paired with ligand. With present computing resources, it is impossible to exhaustively explore the search space this would enumerating all possible distortions of each molecule and all possible rotational and translational orientations of the ligand relative to the protein at a given level of granularity. Most docking programs in use account for flexible ligand, and several are attempting to model a flexible protein receptor in following figure.[15]

Major steps involved in mechanics of molecular docking [14]
Molecular Docking is the process in which the intermolecular interaction between 2 molecules was studied in In-silico. In this process, the Macromolecule is the protein receptor. The micro molecule is the Ligand molecule. So, the Docking process involves the following steps:
Step I - preparation of protein:
Three-dimensional structure of the Protein should be retrieved from Protein data bank (PDB); afterward the retrieved structure should be pre-processed. This should admit removal of the water molecules from the cavity, stabilizing the charges, filling the missing residues, generation the side chains etc. according to the parameters available.
Step II - active site prediction:
After the preparation of protein, the active site of protein should be predicted. The receptor might possess lots of active sites merely the one of the concern should be picked out. Mostly the water molecules and hetero atoms are removed if present.
Step III - preparation of ligand:
Ligands can be retrieved from several databases s such as ZINC, Pub Chem or can be sketched applying Chem Draw tool. While picking out the ligand, the LIPINSKY’S RULE OF 5 should be utilized. Lipinski rule of 5 assists in discerning amongst non-drug like and drug like candidates. It promises high chance of success or failure due to drug likeness for molecules abiding by with 2 or more than of the complying rules. For choice of a ligand allowing to the LIPINSKY’S RULE:
Step IV- docking:
Ligand is docked against the protein and the interactions are analyzed. The scoring function gives score on the basis of best docked ligand complex is picked out[14]
Empirical scoring function of any docking program is
Fitness = vdW + Hbond + Elec
Available Softwares For Docking:
Applications of molecular docking
Molecular docking is a key tool in structural molecular biology and computer-assisted drug design. The goal of ligand protein docking is to predict the predominant binding mode(s) of a ligand with a protein of known three-dimensional structure. Molecular docking is usually performed between a small molecule and a target macromolecule. This is often referred to as ligand– protein docking, but there is growing interest in protein–protein docking. For performing molecular docking, primarily two types of approaches are used. One of the approaches employs computer simulations in which energy profiling is estimated for ligand target docked conformer whereas, second approach utilizes a technique that calculates surface complementary human ligand and target. Molecular docking has a wide variety of uses and applications in drug discovery, including structure–activity studies, lead optimization, finding potential leads by virtual screening, providing binding hypotheses to facilitate predictions for mutagenesis studies, assisting x-ray crystallography in the fitting of substrates and inhibitors to electron density, chemical mechanism studies, and combinatorial library design [10].To determine their tentative binding parameter. This establishes raw data for the rational drug designing for new agents/existing agent with better efficacy and more specificity.[15]
METHODOLOGY
Docking Methodology:
Accession of target protein
The three-dimensional structure of Enoyl acyl carrier protein reductase (InhA)receptor (PDB: 5w07) was downloaded from the RCSB protein Data Bank.[11]
Ligand selection
The chemical structure of Ligand were drawn from chemdraw. Ligand which is present in the pdb format was converted to pdbqt file using Autodocktool to generate atomic coordinates.
Target and ligand optimization
For docking analysis, PDB coordinates of the target protein and Ligand molecule were optimized by Drug Discovery Studio software, respectively (adding missing residues). These coordinates had minimum energy and stable conformation.
Analysis of target active binding sites
The active sites are the coordinates of the ligand in the original target protein grids, and these active binding sites of target protein were analyzed using the Drug Discovery Studio and 3D Ligand Site virtual tools…[12]
Molecular docking analysis
A computational ligand-target docking approach was used to analyze structural complexes of the Enoyl acyl carrier protein reductase (InhA) receptor (target) with ligand molecules in order to understand the structural basis of this protein target specificity. Initially, protein–ligand attraction was investigated for hydrophobic/ hydrophilic properties of these complexes by Autodocksoftware.[13] Finally, docking was carried out by Auto Dock. The energy of interaction of Enoyl acyl carrier protein reductase (InhA)receptor with the Ligand molecules is assigned “grid point.” At each step of the simulation, the energy of interaction of ligand and protein was evaluated using atomic affinity potentials computed on a grid. The remaining parameters were set as default.
Prepared Drugs for Anti-Tuberculosis
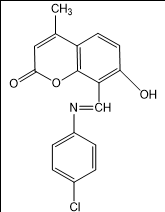
1) E)-8-(((4-chlorophenyl) imino) methyl)-7-hydroxy-4-methyl-2H-chromen-2-one(3a)

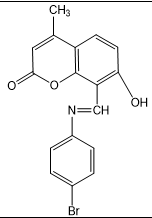
2) E)-8-(((4-bromophenyl)imino)methyl)-7-hydroxy-4-methyl-2H-chromen-2-one(3b)

3) E)-7-hydroxy-8-(((4 hydroxyphenyl)imino)methyl)-4-methyl-2H-chromen-2-one(3c)
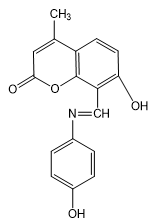
4) (E)-7-hydroxy-4-methyl-8-(((4-nitrophenyl)imino)methyl)-2H-chromen-2-one(3d)
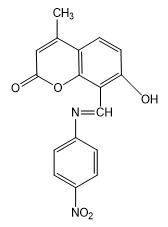
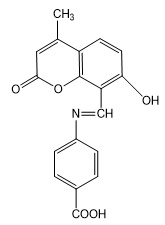
5) E)-4-(((7-hydroxy-4-methyl-2-oxo-2H-chromen-8-yl)methylene)amino)benzoic acid(3e)
RESULTS AND DISCUSSION
Table 1: Physical characterization data of Coumarin Derivatives
|
SL NO |
|
Drug Name |
Molecular weight(g/mol) |
Log-p |
NO of rotatable bonds |
Polymer surface area (Az) |
Molar refractivity (m3/mol) |
|
1 |
3a |
(E)-8-(((4-chlorophenyl)imino)methyl)-7-hydroxy-4-methyl-2H-chromen-2-one |
313.74 |
2.47 |
2 |
62.80 |
88.18 |
|
2 |
3b |
(E)-8-(((4-bromophenyl)imino)methyl)-7-hydroxy-4-methyl-2H-chromen-2-one |
358.19 |
2.61 |
2 |
62.80 |
90.87 |
|
3 |
3c |
(E)-7-hydroxy-8-(((4-hydroxyphenyl) imino)methyl)-4-methyl-2H-chromen-2-one |
295.29 |
1.88 |
2 |
83.03 |
85.19 |
|
4 |
3d |
(E)-7-hydroxy-4-methyl-8-(((4-nitrophenyl) imino)methyl)-2H-chromen-2-one |
324.29 |
1.87 |
3 |
108.62 |
91.99 |
|
5 |
3e |
(E)-4-(((7-hydroxy-4-methyl-2-oxo-2H-chromen-8-yl)methylene)amino)benzoic acid |
323.30 |
1.80 |
3 |
100.10 |
90.13 |
Table 2: Binding energy and bond interaction of Coumarin Derivatives
|
SL NO |
|
Drug Name |
Binding affinity (Kcal/mol) |
RMSD |
Amino acid Interaction |
Type of interaction |
|
1 |
3a |
(E)-8-(((4-chlorophenyl)imino)methyl)-7-hydroxy-4-methyl-2H-chromen-2-one |
-5.15 |
0 |
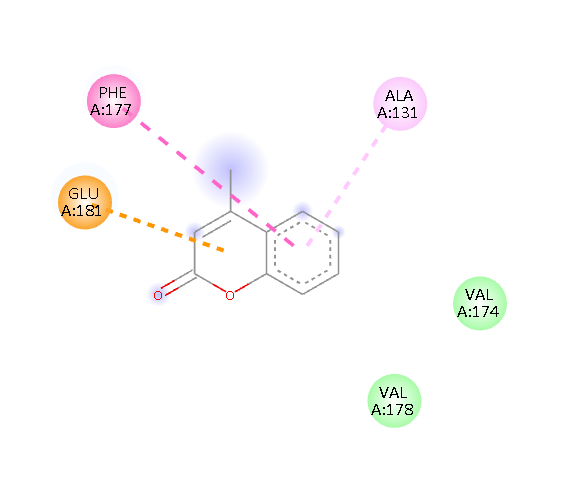
GLU 181 PHE 177 ALA 131 |
Pi-Anion Pi-Alkyl Van der waals Van der waals |
|
2 |
3b |
(E)-8-(((4-bromophenyl)imino)methyl)-7-hydroxy-4-methyl-2H-chromen-2-one |
-3.54 |
0 |
ALA 247 ALA 263 ILE 261 LEU 253 THR A244 |
Pi-Alkyl Van der waals Pi-Alkyl Pi-Alkyl Van der waals Van der waals |
|
3 |
3c |
(E)-7-hydroxy-8-(((4-hydroxyphenyl)imino)methyl)-4-methyl-2H-chromen-2-one |
-4.49 |
0 |
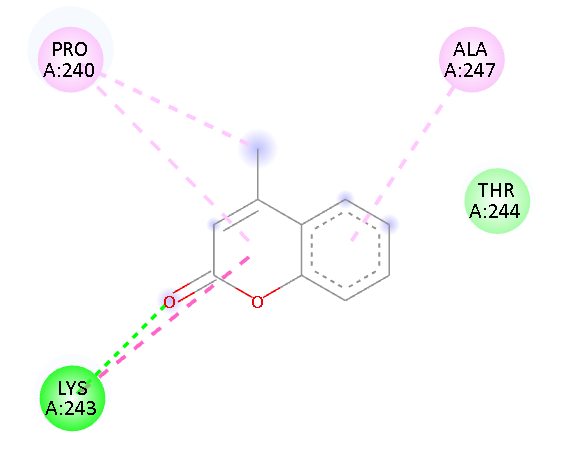
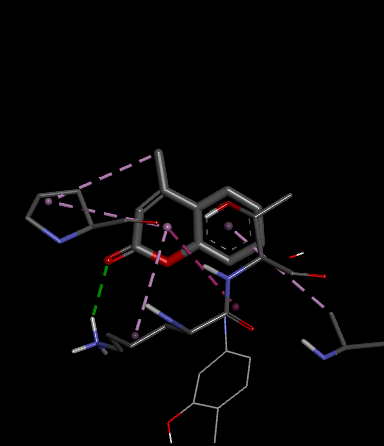
PRO 240 ALA 247 LYS 243 |
Pi-Alkyl Pi-Alkyl H-Bond Van der waals |
|
4 |
3d |
(E)-7-hydroxy-4-methyl-8-(((4-nitrophenyl)imino)methyl)-2H-chromen-2-one |
-5.91 |
0 |
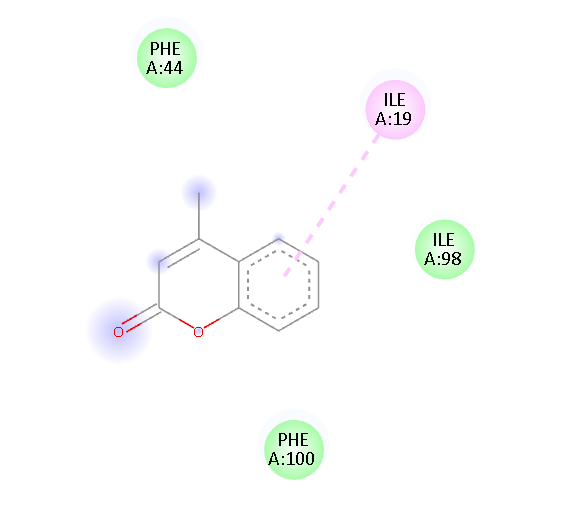
ILE 19 |
Pi-Alkyl Van der waals Van der waals Van der waals |
|
5 |
3e |
(E)-4-(((7-hydroxy-4-methyl-2-oxo-2H-chromen-8-yl)methylene)amino)benzoic acid |
-2.4 |
0 |
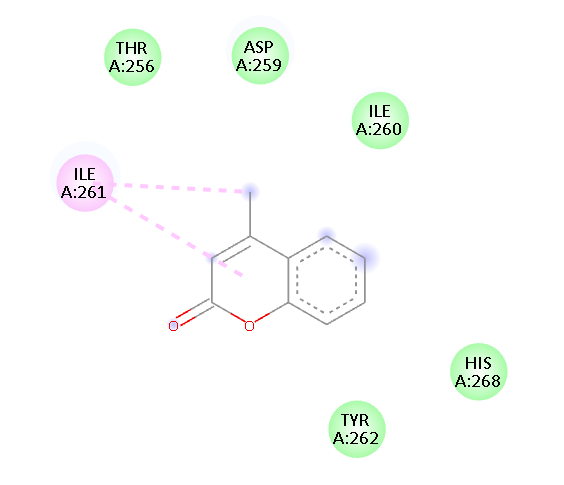
ILE A261 TYR A262 HIS A268 |
Pi- Alkyl Van der waals Van der waals Van der waals Van der waals Van der waals |
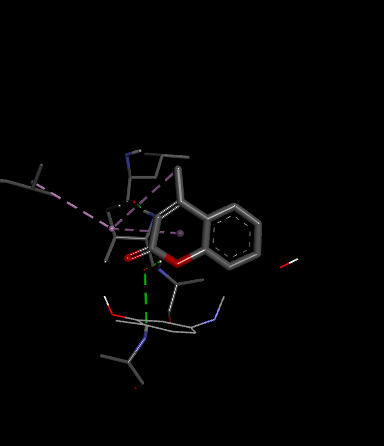
Fig.1 2D Plot Interaction of Ligand molecules with PDB 5w07
|
|
Drug ame |
2d interaction map |
Ligand with 2jx4 |
|
3a |
(E)-8-(((4-chlorophenyl)imino)methyl)-7-hydroxy-4-methyl-2H-chromen-2-one |
|
|
|
3b |
(E)-8-(((4-bromophenyl)imino)methyl)-7-hydroxy-4-methyl-2H-chromen-2-one |

|

|
|
3c |
(E)-7-hydroxy-8-(((4-hydroxyphenyl)imino)methyl)-4-methyl-2H-chromen-2-one |
|
|
|
3d |
(E)-7-hydroxy-4-methyl-8-(((4-nitrophenyl)imino)methyl)-2H-chromen-2-one |
|

|
|
3e |
(E)-4-(((7-hydroxy-4-methyl-2-oxo-2H-chromen-8-yl)methylene)amino)benzoic acid |
|
|
Table 4: Lipinski rule of 5 of Coumarin Derivatives
|
|
Drug name |
Structure |
MW(g/mol) |
Log-p |
HBA |
HBD |
violation |
|
3a |
(E)-8-(((4-chlorophenyl)imino)methyl)-7-hydroxy-4-methyl-2H-chromen-2-one |
|
313.74 |
2.47 |
4 |
1 |
0 |
|
3b |
(E)-8-(((4-bromophenyl)imino)methyl)-7-hydroxy-4-methyl-2H-chromen-2-one |
|
358.19 |
2.61 |
4 |
1 |
0 |
|
3c |
(E)-7-hydroxy-8-(((4-hydroxyphenyl)imino)methyl)-4-methyl-2H-chromen-2-one |

|
295.29 |
1.88 |
5 |
2 |
0 |
|
3d |
(E)-7-hydroxy-4-methyl-8-(((4-nitrophenyl)imino)methyl)-2H-chromen-2-one |

|
324.29 |
1.87 |
6 |
1 |
0 |
|
3e |
(E)-4-(((7-hydroxy-4-methyl-2-oxo-2H-chromen-8-yl)methylene)amino)benzoic acid |

|
323.30 |
1.80 |
6 |
2 |
0 |
We design a series of coumarin derivatives and screened for molecular docking studies using the receptor InhA (PDB ID: 5W07) using the Autodock tools. InhA receptor is part of the type II fatty acid biosynthesis pathway in M. tuberculosis. This pathway is essential for producing mycolic acids, which are unique to mycobacteria and vital for constructing the bacterial cell wall. We designed a novel derivatives of coumarin to explore their binding mode with InhA receptor. So the binding affinity and amino acid residues with designed compounds and standard drug Isoniazid interacted are presented in the Table no- 2 and fig-1. Among the designed compounds, compound with electron withdrawing group substituted like Chloro (Cl) 3a and Nitro (NO2) 3d exhibited most favorable binding affinity towards the targeted protein by interaction with different amino acid residues and shows the strong interaction and potential inhibitory activity as compare to compounds containing electron donating groups.
CONCLUSION
In this study, a series of novel coumarin derivatives (3a–3e) were designed and evaluated through molecular docking to explore their potential as anti-tuberculosis agents. Among the designed compounds, compound 3d exhibited the most favorable binding affinity towards the target protein, indicating strong interaction and potential inhibitory activity. The docking results suggest that compound 3d effectively occupies the active site of the mycobacterial enzyme, forming key interactions that are crucial for its anti-TB action. These finding position compound 3d as a promising lead candidate for further in vitro and in vivo studies aimed at developing effective therapeutics against tuberculosis.
REFERENCES


Veeresh K.*, Arunakumar, Ashwini, Supriya B. M., Santhoshkumar, Manappa M., Design and Molecular Docking Studies of Novel Coumarin Derivatives Against Tuberculosis, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 1398-1417. https://doi.org/10.5281/zenodo.15854249
 10.5281/zenodo.15854249
10.5281/zenodo.15854249