Krupanidhi College of Pharmacy, Bangalore.
Hutchinson-Gilford progeria syndrome (HGPS) is a sporadic genetic disorder characterized by dramatic, accelerated aging in children. Primarily caused by mutations in the LMNA gene, which encodes the lamin A protein, HGPS results in the production of an abnormal protein known as progerin. This defect disrupts the structure of the cell nucleus, leading to premature aging symptoms that manifest in infancy or early childhood, including growth failure, wrinkled skin, hair loss, and severe cardiovascular issues reminiscent of those seen in aging populations. The syndrome affects approximately 1 in 4 million live births, underscoring its rarity and the unique challenges faced by affected families [1][2][3]. Clinically, HGPS is characterized by a distinctive set of symptoms, including joint stiffness, cardiovascular complications, and delayed dental development, while cognitive function remains largely intact. As the condition progresses, individuals often experience significant health complications that can lead to a markedly shortened lifespan, with average life expectancy being around 14.5 years [1][4]. The most common causes of mortality are heart failure and stroke, largely resulting from severe atherosclerosis [1][5][4]. Notably, although there is currently no cure, recent advancements in pharmacological treatment, particularly with the approval of lonafarnib, have shown promise in extending life expectancy and improving quality of life for those affected [1][6][7]. The rarity of HGPS, coupled with its severe manifestations, has led to considerable attention in the medical and scientific communities. Research efforts are focused on unraveling the molecular mechanisms of the syndrome and developing effective therapeutic strategies, including gene therapy and innovative drug treatments. Collaborative initiatives, particularly by organizations like the Progeria Research Foundation, aim to enhance understanding and foster research advancements, as well as to improve clinical outcomes for individuals with HGPS globally [8][6]. However, ongoing challenges such as high treatment costs and the need for comprehensive care remain prominent issues, necessitating a multidisciplinary approach to management and support [9][6].
Symptoms
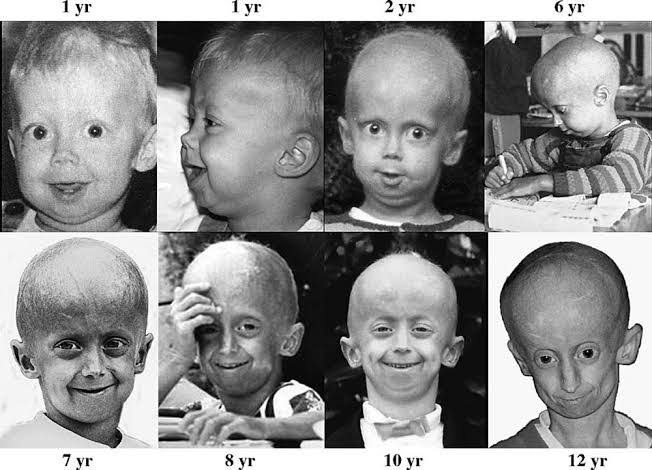
Hutchinson-Gilford progeria syndrome (HGPS) is characterized by a distinct array of clinical symptoms that resemble accelerated aging, manifesting predominantly during infancy or early childhood. Common symptoms include growth failure and short stature, where children typically fall below the third percentile for height and weight[1][2]. As the condition progresses, affected individuals exhibit wrinkled, aged-looking skin, hair loss, including loss of eyebrows and eyelashes, and visible veins on the scalp and extremities[3][2][10]. Facial features are also notably altered, with individuals displaying a narrow face, a prominent forehead, a small jaw, and a distinctive beaked nose[3][2][10]. The high-pitched voice and thin, spotty skin further contribute to the characteristic appearance of children with progeria[3][2]. Joint issues such as stiffness and pain, particularly in the hips and knees, are common, often leading to joint dislocations and requiring careful management through physical therapy[11][2]. Furthermore, severe cardiovascular complications emerge as a significant concern, with affected individuals experiencing progressive heart and blood vessel diseases, including atherosclerosis, which can result in life-threatening events such as heart attacks and strokes[3][2][10][12]. Other health issues include delayed tooth formation, skeletal abnormalities, and metabolic disorders that typically worsen with age[2][13]. Despite these challenges, cognitive development remains largely unaffected, allowing children with HGPS to maintain social and intellectual growth[3][2].
Causes
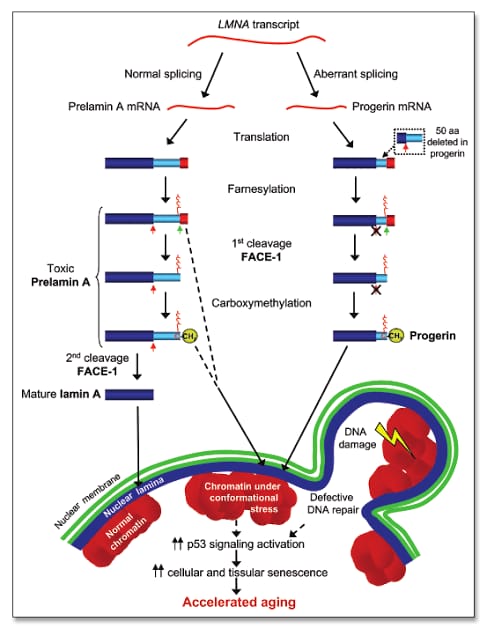
Hutchinson-Gilford Progeria Syndrome (HGPS) is primarily caused by mutations in the LMNA gene, which encodes the lamin A protein, a critical component of the nuclear envelope that maintains nuclear integrity and stability[14][10]. Approximately 80% of HGPS cases are linked to a specific point mutation known as G608 (GGC→GGT) within exon 11 of the LMNA gene, which leads to the production of an abnormal form of lamin A called "progerin"[15][2]. This defective protein causes nuclear instability, resulting in chronic cellular damage and premature cell death, which are hallmarks of the syndrome[14][16]. The mechanisms underlying HGPS are thought to involve accelerated telomere shortening and disruptions in gene expression, contributing to various age-related complications, particularly cardiovascular issues. As a result, children with progeria experience symptoms that mimic aging, such as skin wrinkling, joint stiffness, and atherosclerosis, which manifests as early-onset cardiovascular disease, heart failure, and stroke. Notably, progeria often arises from de novo mutations, meaning that affected individuals typically do not have a family history of the disorder, which significantly contributes to its rarity, occurring in approximately 1 in 4 million live births. In addition to HGPS, the LMNA gene is implicated in a range of disorders collectively referred to as laminopathies, which can exhibit varying clinical phenotypes associated with accelerated aging and other systemic complications [5].
Diagnosis
Hutchinson-Gilford Progeria Syndrome (HGPS) is primarily diagnosed through clinical observation of characteristic signs and symptoms, such as aging skin, hair loss, and growth abnormalities. Health care providers often suspect progeria based on these clinical features and a thorough physical examination, which includes measuring height and weight, testing hearing and vision, and monitoring vital signs, including blood pressure[18][19]. To confirm a diagnosis of HGPS, a genetic test is available that specifically examines mutations in the LMNA gene, which is responsible for the production of lamin A, a protein crucial for nuclear structure stability. In cases where genetic testing is utilized, it has been noted that the majority of patients diagnosed with HGPS have classic variants of the LMNA mutation, particularly the c.1824C>T pathogenic variant. While genetic confirmation is essential for accurate diagnosis, it is also acknowledged that some patients may be categorized based solely on clinical features, particularly in instances where genetic testing is not performed or results are inconclusive. This approach may lead to variations in diagnostic accuracy, especially given the heterogeneous nature of progeroid laminopathies and the existence of atypical variants[13]. Therefore, an integrated approach combining clinical evaluation and genetic analysis is crucial for diagnosing HGPS effectively and ensuring appropriate patient management[6][3].
Treatment
Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare genetic disorder characterized by accelerated aging in children, resulting in significant health challenges. The treatment landscape for HGPS has evolved significantly, with various therapeutic strategies emerging to address the condition.
Supportive Care
In addition to FTIs, supportive care plays a crucial role in managing complications associated with HGPS. This may include physical and occupational therapy to address joint stiffness and daily living challenges, nutritional support to ensure adequate caloric intake, and medications for symptom management, such as statins or blood thinners[19]. Multidisciplinary approaches are essential, involving pediatric cardiology, genetics, and specialized teams to monitor and manage cardiovascular health, which is particularly impacted by progeria[9].
Pharmacological Interventions
The primary pharmacological treatment for HGPS is Zokinvy (lonafarnib), a farnesyltransferase inhibitor (FTI) that prevents the abnormal protein progerin from causing cellular damage by inhibiting its association with the nuclear membrane
Future Directions
Research is ongoing to identify and develop new treatment modalities, including gene therapy aimed at directly addressing the underlying genetic mutation causing HGPS. Emerging strategies also involve progerin-targeting drugs and advanced CRISPR-based gene therapies, which hold promise for more precise interventions in the future.
Trend Of Hutchinson-Gilford Syndrome (2025 to 2035)
Prognosis
Hutchinson-Gilford progeria syndrome (HGPS) is characterized by a markedly shortened lifespan, with an average life expectancy of approximately 14.5 years; however, some individuals may succumb to complications as early as 6 years old, while a few may live into their early 20s. The most common causes of mortality in HGPS patients include heart failure and stroke, primarily resulting from severe atherosclerosis and cardiovascular complications that resemble those typically seen in elderly individuals [5][1][4]. Clinical observations indicate that the trajectory of growth and the emergence of complications begin around the age of one, with notable deviations from typical growth patterns by this age. Cardiovascular issues, including vascular damage and arrhythmias, are prevalent and often manifest during childhood, necessitating close monitoring as patients age. Moreover, renal complications, such as kidney failure, have been observed in long-term survivors, highlighting the need for vigilant renal function assessment as patients approach late-stage disease [13]. Emerging treatments, particularly the farnesyltransferase inhibitor lonafarnib, have shown promise in extending life expectancy by an average of 2.5 years. Continued advancements in therapeutic options, along with multidisciplinary care approaches, aim to improve both the quality of life and longevity of individuals with HGPS. Future research is essential to elucidate long-term complications and develop more effective treatment protocols, as current therapies focus predominantly on managing cardiovascular and renal health [13][14][9].
Future Directions
Looking ahead, the research landscape for HGPS is poised for significant growth, fueled by advancements in biotechnology, government funding for orphan drug development, and an increased focus on personalized medicine. Continued exploration of combination therapies, along with the application of emerging genetic and cellular therapies, promises to further evolve the therapeutic options available for individuals with HGPS. The integration of genomic medicine into treatment strategies is anticipated to enhance patient outcomes, ultimately leading to a more nuanced understanding of the disease and its management [9].
Overview of Hutchinson-Gilford Progeria Syndrome (HGPS)
Hutchinson-Gilford progeria syndrome (HGPS) is a rare genetic disorder characterized by accelerated aging, primarily caused by mutations in the LMNA gene, which encodes lamin A. This mutant form of lamin A, known as progerin, disrupts nuclear architecture and leads to cellular senescence and various age-related phenotypes in affected individuals. Current research efforts focus on understanding the molecular mechanisms that underpin the pathophysiology of HGPS and identifying viable therapeutic targets[6][21].
Advances in Therapeutics
Recent advancements in the development of therapeutic interventions for HGPS have been significant. Researchers are exploring traditional small-molecule drugs, biologics, RNA therapeutics, and gene-editing approaches such as CRISPR. These modalities have shown promise in preclinical models, including HGPS cell lines and mouse models, leading to better insights into disease progression and potential treatment options. Notably, farnesyltransferase inhibitors (FTIs) like lonafarnib have emerged as a leading treatment, slowing disease progression by targeting progerin accumulation. Furthermore, innovative animal models are being developed to address critical questions in translational research, thereby enhancing the prospects for effective therapies [6].
Epidemiological Insights
Epidemiological studies have begun to provide a clearer picture of the prevalence and clinical characteristics of HGPS. Recent research in Japan indicates that the national prevalence of HGPS is comparable to that in the U.S. and China, underscoring the importance of international comparisons in refining care strategies for affected individuals[13]. These studies also emphasize the need for comprehensive support resources to aid patients and families throughout their lives, particularly in the context of long-term care and management[13].
Collaborative Efforts in Research
The Progeria Research Foundation (PRF) plays a pivotal role in fostering collaboration among scientists and clinicians, funding numerous research projects aimed at HGPS. Since its inception, PRF has allocated over $7.7 million to support 71 research grants across multiple countries, illustrating a robust commitment to advancing the understanding and treatment of this rare disorder. Furthermore, the global landscape of HGPS research is increasingly characterized by collaborative efforts that bridge basic science and clinical application, thereby enhancing the potential for innovative treatment solutions[6][9].
Notable Cases
Overview of HGPS Cases in Japan
A recent study presented a comprehensive examination of Hutchinson-Gilford Progeria Syndrome (HGPS) through the analysis of several notable cases in Japan. This cohort included eight patients with confirmed HGPS diagnoses, representing a balanced sex distribution of four males and four females[13]. Genetic analysis revealed that seven of these cases exhibited classic variants of the disease (c.1824C>T), while one case presented with an atypical variant (c.1968+1G>A). Additionally, two further patients were clinically diagnosed without genetic confirmation, highlighting the diagnostic challenges associated with this ultra-rare condition.
Categories of Patients
The study categorized the patients into two distinct groups based on the reliability of their diagnoses. The confirmed group consisted of Cases 1 to 8, all exhibiting HGPS, while Cases 9 and 10 were designated as unconfirmed due to their reliance on clinical diagnosis without genetic validation. This classification underlines the need for stringent diagnostic protocols to enhance the accuracy of HGPS diagnoses.
Clinical Insights and Future Directions
While the primary focus of the study was on genetic and clinical characteristics, it also indicated that social and psychological factors affecting quality of life were not systematically assessed. These elements represent significant burdens for patients and their families and underscore the necessity for future multicenter studies to encompass a more holistic understanding of HGPS and related laminopathies. Moreover, the introduction of standardized treatments, such as Lonafarnib, has emerged as a critical aspect of improving patient outcomes, especially considering its approval for individuals aged one year and older[13]. Thus, ongoing research and international collaboration are vital for advancing knowledge and treatment options for HGPS patients globally.
REFERENCES

Ayan Sarkar*, Detailed Case Study On: Hutchinson-Gilford Progeria Syndrome, A Rare Genetic Disorder Characterized by the Rapid Appearance of Aging in Children, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 2501-2508. https://doi.org/10.5281/zenodo.16076554
 10.5281/zenodo.16076554
10.5281/zenodo.16076554
