Dr. Uttamrao Mahajan College of Pharmacy, Chalisgaon, Maharashtra, India.
Aim: The main purpose of the research work is to develop an effective, sensitive, provident and uncomplicated reverse phase HPLC technique for the estimation of Tofacitinib in Tofacitinib tablets dosage form. Study Design: HPLC based grounded quantification studies. Methodology: Estimation of Tofacitinib in Tofacitinib tablet formulation. The separation was set up by using a stationary phase Kromasil C18 (150 x 4.6 mm, 5µ) and the mobile phase comported of pH 4.0 phosphate buffer and acetonitrile in the rate of (80:20 volume/volume). The flow rate was 1.5 mL/min. Tofacitinib was detected using UV sensor at the wavelength of 215 nm. composed of pH 4.0 phosphate buffer and acetonitrile in the rate of (80:20 volume/volume). The flux rate was 1.5 mL/min. Column temperature 25°C and sample temperature 25°C and injection volume 20µL, performance time was 20 moment. Results And Conclusion: The citified technique was validated for numerous parameter as per ICH guidelines like accuracy, precision, linearity, specificity, system suitability, result stability and robustness. The results attained were within the undertaking criteria. So, it can be concluded that the formulated system is easy, specific, cost-efficient, sustainable, and safe and can be successfully employed for the procedure analysis of Tofacitinib in tab and pharmaceutical dosage forms.
Pharmaceutical Drug Analysis
Modern drug analysis deals with qualitative and/or quantitative estimations (constitutional quantification), using spectroscopy and chromatography techniques. Pharmaceutical drug analysis can be defined as the science and art of determining the purity of a substance in bulk, from formulations/biological matrices, and in presence of the impurities. The pharmaceutical analysis aids in the identification of pharmaceuticals, foreign contaminants in medications, and structural elucidation of drug-related substances. The examination of drug compounds employs the use of analytical methods with classical or instrumental approaches for routine analysis. Analytical procedures developed using UV-spectrophotometer, HPLC, HPTLC, and GC have a wide range of applications in ensuring the quality and quantity of pharmaceutical drugs, raw materials, and formulations. These procedures are simple to use, accurate, precise, and reproducible and thus form an important aspect in pharmaceutical drug investigation.
Important factors which must be considered while selecting an analytical method.
Advantages of Instrumental Methods
Chromatography
Chromatography has the unique and wide role in pharmaceutical analysis. It helps in precise separation of the drug compounds, their analysis and in purification. It requires very low sample volumes and works in wide range of samples. Thus, it plays important role in analysis of pharmaceutical analysis, “May be defined as a method of separating a mixture of components into individual components through equilibrium distribution between two phases.
It is based on the difference in the rate at which the components of mixture move through a porous medium (called stationary phase) under the influence of some solvent or gas (called moving phase)
The IUPAC has defined chromatography as “A Analytical technique of separation of mixture in which the components to be separated are dispersed between two phases, one of which is stationary while the other moves in a certain direction”
Chromatography is The Chromatographic Method of Separation, which involve the following steps
majorly classified in two types that are partition and adsorption chromatography
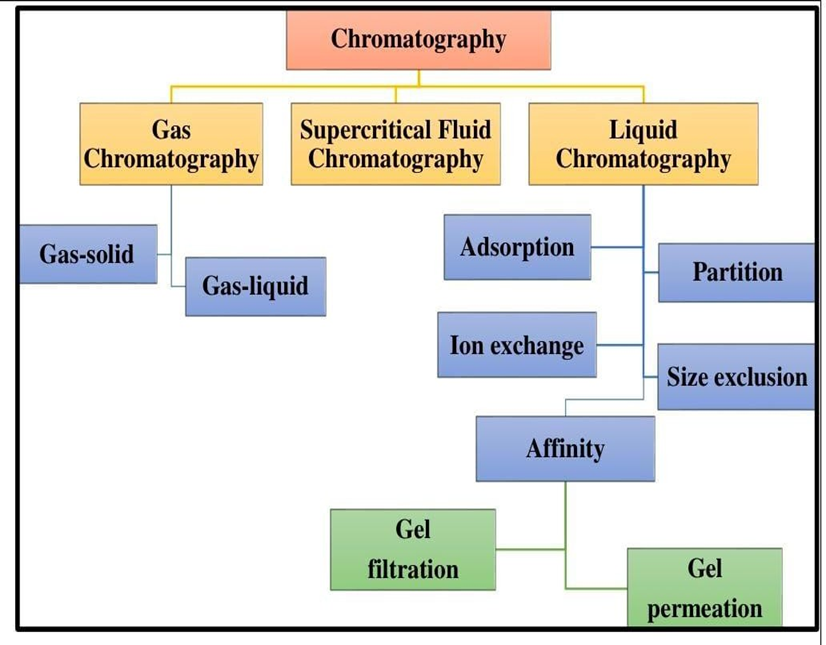
Classification of chromatography technique
High-performance liquid-chromatography (HPLC)
High-performance liquid chromatography (HPLC) is a key technique for separating and analyzing chemical mixtures. It utilizes conventional detectors that rely on the analyte's refractive index, UV, fluorescence, and electrochemical properties . HPLC is the preferred tool for analysis in the rapidly advancing field of pharmaceutical research, and efforts have been ongoing to enhance the efficiency of chromatographic technique development.
Types of HPLC
Advantages:
Disadvantages:
Drug Profile and Literature Survey Tofacitinib
Janus kinases are a type of phosphotransferase, which are enzymes that become active when cytokines bind to their receptors. Janus kinase inhibitors provide a new strategy for treating immune and inflammatory diseases. Tofacitinib is an orally administered, targeted synthetic small molecule that acts as an inhibitor of Janus kinases (JAKs) and has received FDA approval. Tofacitinib chemically known as 3-[(3R, 4R) - 4 -methyl-3- [methyl (7H-Pyrrolo [2, pyrimidine-4yl) amino] piperidin-1-yl]-3oxopropanenitrile. It is an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis.
Its mechanism of action primarily involves the inhibition of the non-receptor tyrosine kinase JAK enzymes, with a preference for JAK-1 and JAK-3.ymes. Tofacitinib is indicated for adult patients with rheumatoid arthritis who have not responded to or have developed intolerance to one or more disease-modifying antirheumatic drugs (DMARDs) and who have an active moderate to severe disease course. Tofacitinib can be used in conjunction with first-line therapy such as methotrexate (MTX) or other conventional DMARDs, or it can be used as a standalone treatment.
Degradation of tofacitinib follows apparent first-order kinetics. In order to maximize stability of the drug, ionic strength and temperature should be minimized, with an optimal range pH between 2.0 and 5.0.
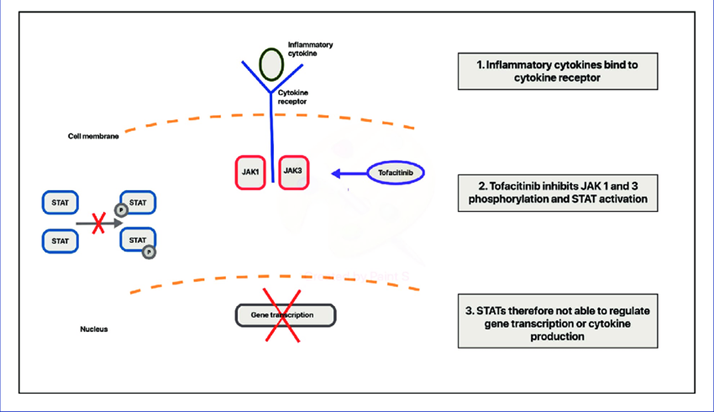
Mechanism of Action of Tofacitinib
Drug Profile
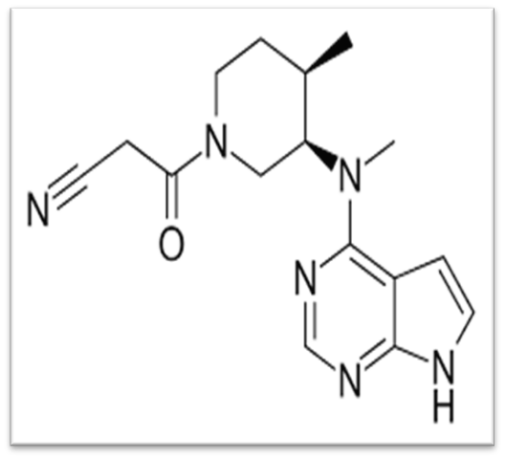
Figure 2. Chemical Structure of Tofacitinib
Molecular Formula : C16H20N6O
Molecular Weight : 312.37g/mol
Chemical Name : 3-((3R, 4R)-4-methyl-3-(methyl (7H-pyrrolo [2, 3-d] pyrimidin-4-yl) amino) piperidin-1-yl)-3-oxopropanenitril
Melting Point : 198-202° C
Solubility Category: N, N-Dimethylacetamide
Pka: 5.2
Review of Literature for Tofacitinib
A comprehensive review of literature has been performed to determine the availability of analytical reports for determining Tofacitinib in pharmaceutical and biological matrices. The detailed literature survey for cited analytes is as follows
Analysis of Tofacitinib using HPLC includes;
Kaleswararao et. al; A reversed-phase RP-HPLC method for Tofacitinib was developed using a C18 column (150 x 4.6 mm), 3 μm as the stationary phase. The mobile phase comprised 0.4 % perchloric acid and acetonitrile in the ratio of 85:15 % v/v. The mobile phase flow rate was maintained at 1.2 mL/min, with the temperature set at ambient level. The column temperature was maintained at 40ºC to get early elution of analyte and sample cooler temperature at 10 ºC. Texwipe swabs with part number TX761D have been selected as swab sticks for sample collection as the risk of fibre interference is absent. [33]
Sankar as et. al; A novel RP-HPLC method has been developed and validated for the estimation of Tofacitinib in its tablet dosage form. The analysis was performed using an isocratic system on a C18 column with the dimensions of (150 mm X 4.6 mm) and 5 micron particles at ambient temperature. . The mobile phase consisted of methanol: water in a ratio of 90:10% (v/v), with a flow rate maintained at 0.8 ml/min. Detection was conducted at a wavelength of 285.9 nm. Validation of the method was conducted in accordance with ICH guidelines, covering system suitability, linearity, accuracy, and precision. The linear range for TDF was determined to be 50-150 µg/mL, with accuracy and precision well within acceptable limits. [34]
Sivaprasadu et. al; A new method was developed for the estimation and validation of TFC in its pure form and pharmaceutical dosage form using RP-HPLC. mobile phase composed of phosphate buffer (pH 7.0) and acetonitrile (45:55, %v/v) at a flow rate of 0.5 mL/min under isocratic elution. Analytes were monitored via a UV detector at 210 nm and with the column oven temperature at 30°C for a 20-min analysis. . Precision (%RSD) for Impurity-A and Impurity-B and tofacitinib met specifications at 2.4%, 0.8%, and 0.0%, respectively. Accuracy ranged from 86% to 100% for impurities, with LOD at 0.03% and LOQ at 0.05%–0.06%. Correlation coefficients exceeded 0.999 for impurities and tofacitinib citrate. Solution stability was confirmed for 24 h at room temperature . Validation of the method was performed following various parameterssuch as accuracy, precision, linearity, and specificity according to the ICH guidelines.[35]
Ozbay et. al; An analytical method was developed for Tofacitinib and one of its impurities, requiring synthesis and characterization of the impurity. s. The newly developed method exhibits simplicity, specificity, precision, and sensitivity. Experimental procedures utilized a Shimadzu Prominence 20A HPLC system equipped with a Inertsil ODS 3V C18 column (5µm particle size, 4.6 X 250 mm dimensions). The mobile phase, consisting of 0.05M ammonium acetate buffer at pH 5.0 and acetonitrile (65:35 v/v) in isocratic mode with a flow rate of 1.0 mL/min, facilitated accurate detection of the Tofacitinib peak at 230 nm wavelength. Comprehensive validation, including assessments of linearity, accuracy, precision, and robustness, was conducted in accordance with ICH requirements.
Aim
The aim of this research is to develop and validate a rapid, simple, To Develop Stability indicating Method for determination of Tofacitinib in Tofacitinib tablet dosage Form by using RP HPLC and UV Spectroscopy.
OBJECTIVE
Plan of work
The main intention of the research work is to develop and effective, sensitive, economical and reverse phase HPLC technique for the estimation of Tofacitinib in Tofacitinib tablets dosage form.
|
Mobile Phase ( Buffer) |
Instead of various buffer we can uses acidic acid,formic acid Phosphate buffer, ammonium acetate. |
Forced Degradation Study :- A forced degradation study (FDS) in HPLC (High-Performance Liquid Chromatography) is crucial for drug development, intentionally stressing a drug substance (API) with acid, base, heat, light, and oxidation to generate impurities, proving the HPLC method is stability-indicating, meaning it separates API from degradants, identifying potential degradation pathways, and ensuring method specificity for regulatory submission. The primary purpose is to help develop and validate a stability-indicating method using High-Performance Liquid Chromatography (HPLC) that can accurately separate, detect, and quantify the drug substance and its various degradation products.
Method For RP-HPLC
Development of Method
Validation of Method
Methodology
Development and Validation Method
RP- HPLC Method
Analytical-grade Potassium dihydrogen phosphate, Orthophosphoric acid, Acetonitrile, Hydrochloric acid, Sodium hydroxide, Hydrogen peroxide and water, reagents and chemicals.
Agilent HPLC model:1260 with DAD, Bandelin ultrasonic bath, pH Meter (Thermo Orion Model) and Analytical Balance (Metller Toledo Model) were used in the present assay.
Weighed accurately 1.36 g of potassium dihydrogen orthophosphate into 1000 mL of water sonicated to dissolve and mix well then pH was adjusted to 4.0 with ortho phosphoric acid solution. Filtered through 0.45 µm membrane filter.
Prepared a mixture of pH 4.0 phosphate buffer solution and acetonitrile in the proportion of 80:20 (%volume/volume) mixed well and sonicated to degas.
Prepared a mixture of irrigate and cyanomethane in the proportion of 50:50 (%volume/volume) mixed well and sonicated to degas.
Weighed correctly and transferred 25.7 mg of Tofacitinib working standard into a 50 mL volumetric thermos sonicated for 2 minutes to dissolve the contents and made upto the quantity with diluent. Further diluted this solution 5 mL in to 50 mL volumetric thermos and made up the quantity with diluent and mixed well. (The concentration of the standard solution containing, Tofacitinib 50 ppm)
Weighed accurate 10 tablets (Tofacitinib tablets 5 mg) and transferred (equivalent to 50 mg of Tofacitinib) into 200 mL volumetric flask, added 120 mL of diluent and sonicated for 30 minutes, with intermediate shaking, cool to room temperature and diluted to quantity with diluent and mixed well. Filtered the solution through 0.45 µm PVDF syringe filter. Further diluted 5.0 mL of the filtrate solution in to 25 mL volumetric thermos, diluted to quantity with diluent, and mixed well. (The concentration of the solution contains 50.0 µg/mL of Tofacitinib).
Weighed correctly 5 tablets (Tofacitinib tablets 10 mg) and transferred (equivalent to 50 mg of Tofacitinib) into 200 mL volumetric thermos, added 120 mL of diluent and sonicated for 30 minutes, with intermediate shaking, cool to room temperature and diluted to quantity with diluent and mixed well. Filtered the solution through 0.45 µm PVDF needle filter. Further diluted 5.0 mL of the filtrate solution in to 25 mL volumetric thermos, diluted to quantity with diluent, and mixed well. (The concentration of the solution contains 50.0 µg/mL of Tofacitinib).
Weighed correctly and transferred placebo powder (equivalent to 50 mg of Tofacitinib) into 200 mL volumetric thermos, added 120 mL of diluent and sonicated for 30 minutes, with intermediate shaking, cool to room temperature and diluted to measure with diluent and merge well. Filtered the solution through 0.45 µm PVDF needle filter. Further diluted 5.0 mL of the filtrate solution into 25 mL volumetric thermos, diluted to quantity with diluent and mixed well. (The concentration of the solution contains 50.0 µg/mL of Tofacitinib).
Chromatographic analysis was executed on Kromasil C18 (150 x 4.6 mm, 5µ) mobile phase consisting of pH 4.0 phosphate buffer and acetonitrile in the proportion of (80:20 volume/volume). The run velocity was 1.5 mL/min, the column oven temperature was 25°C and the sampler cooler temperature was 25°C, the inoculation volume was 20 μL, and detection was implemented at 215 nm using a photodiode array detector (PDA).
Method Development
To develop a appropriate and robust HPLC technique for the determination of Tofacitinib in Tofacitinib in tablets dosage form, different mobile phases were employed to achieve a good peak shape. The technique development was started with Hypersil BDS C18 (150x4.6mm, 5µm) with the following different mobile phase compositions like 0.1% orthophosphoric acid buffer and acetonitrile in the proportion of 85:15 volume/volume. It was observed that when Tofacitinib was injected, higher retention time and zenith tailing were not acceptable. The column stationary phase was not suitable for the component. For the subsequent trial change the column from Hypersil BDS to Kromasil C18. The compound Tofacitinib was eluted at blank volume, and the peak shape was inadequate.
For the subsequent trial the mobile segment consisted of pH 4.0 phosphate buffer and acetonitrile in the proportion of 80:20 volume/volume respectively, flow rate 1.5 mL/min, column temperature 25°C and sampler cooler maintained 25°C. UV detection was performed at 215nm. The compound Tofacitinib was eluted at 10.30 minutes and the peak shape was establish to be good. The chromatogram of Tofacitinib standard using the proposed method . System suitability results of the technique are presented.
Validation Method
The standard solution was organized as per the test technique, infused into the HPLC system six times, and evaluated the % RSD for the vicinity responses.The data were shown in Table 3.The relative standard deviation of six replicates standard solution consequences were establish to be within the specification limit i.e.0.16%.
The linearity of an analytical technique is its ability to obtain test results which has a definite mathematical relation to the concentration of the analyte. The linearity of response for Tofacitinib was determined in the range of 50% to 150% (24.88-74.64 µg/mL for Tofacitinib). The calibration curve of the analytical technique was assessed by plotting concentration versus peak area and represented graphically. The correlation coefficient [r2] was initiate to be 1.000. Therefore, the HPLC technique was established to be a linear standard curve that was calculated and to demonstrate the linearity of the present technique. From the data obtained which is given in Table 5 and the technique was established to be linear within the proposed range.
The accuracy of the test technique was demonstrated by preparing recovery samples of Tofacitinib at 50% to 150% of the target concentration level. The recovery samples were organized in triplicate arrangements on Tofacitinib API spiked to placebo and analyzed as per the recommend technique for apiece attentiveness level except 50% and 150%. The above samples were chromatographed, and the percentage recovery of apiece sample was calculated for the amount added. Evaluated the exactitude of the recovery at apiece level by computing the relative standard deviation of six preparations for 50% and 150% level recovery samples consequences. The data obtained which given in Table 6 and the technique was established to be accurate.
To authenticate the technique robustness the chromatographic performance at distorted circumstances was assessed evaluated to the ostensible conditions of the technique. The standard solution was infused at each of the following altered circumstances. The technique is robust for altered like flow rate, column oven temperature, pH variation, and the organic phase of the mobile phase.
Executed the filter validation for sample solution, one piece of the solution was centrifuged, and the other piece of the solution was filtered through 0.45 µm PVDF and 0.45 µm Nylon filters. Filter validation parameters were established. Based on the above consequences and observations 0.45 µm PVDF filterers are suitable for filtration.
DISCUSSION
RP-HPLC technique for inference of Tofacitinib in Tofacitinib tablets dosage form was urbanized and authenticated as per ICH instructions. A simple, accurate and reproducible reverse phase HPLC technique was urbanized for the connection of Tofacitinib in Tofacitinib tablets dosage form. The optimized technique consists of a mobile segment consisting of pH 4.0 phosphate buffer and acetonitrile in the proportion of (80:20 volume/volume) with Kromasil C18 (150 x 4.6mm, 5µ) column. The retention time of Tofacitinib was established to be 10.30 minutes. The urbanized technique was authenticated as per ICH Q2A (R1) guideline.
CONCLUSION
The urbanized technique was authenticated for various parameters as per ICH guidelines like accuracy, precision, linearity, specificity, system suitability, solution stability and robustness. The result obtained were within the acceptance criteria. So, it can be concluded that the urbanized technique is simple, precise, cost-effective, eco-friendly, and safe and can be successfully employed for the regular analysis of Tofacitinib in bulk and pharmaceutical dosage forms.
REFERENCES


Swati Mahajan, Habiburrahman Shaikh, Dr. Mrunal Shirsat, Development and Validation of Stability Indicating RP-HPLC Method for Quantitative Estimation of Tofacitinib in Tofacitinib Tablets Dosage Form, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 182-192. https://doi.org/10.5281/zenodo.18458165
 10.5281/zenodo.18458165
10.5281/zenodo.18458165
