
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutics, Malla Reddy College of Pharmacy, Hyderabad, 500100.
The formulation of poorly soluble compounds for oral delivery is the most frequent and greatest challenges to formulation scientists in the pharmaceutical industry today. There are many drugs for which dissolution poses a challenge for formulating oral dosage forms leading to bioavailability problems. The drug selected for the present study is Nifedipine belonging to BCS class II. Solubility and hence the dissolution rate of these drugs were enhanced by solvent evaporation technique. The main objective of the study was to enhance the dissolution of Nifedipine, a poorly water-soluble drug by solvent evaporation technique using different carriers and to study the effect of each carrier on the in vitro dissolution profile. The formulations were optimized in the preliminary trials by using various ratios of different carriers like croscarmellose sodium, sodium starch glycolate and ?-Cyclodextrin Resultant formulations were evaluated using FTIR, SEM and in vitro dissolution. Solid dispersion with cyclodextrin 1:5 ratio gave good release when compared to pure drug and physical mixtures. The optimized dispersion was formulated in to oral dispersible tablets by using kyron T-314, cros povidone as disintegrants and was evaluated for friability, hardness, weight variation, disintegration and invitro dissolution. The oral dispersible tablets prepared with kyron T-314 as disintegrant gave good disintegration.
The oral route is the most preferred route of drug administration due to its convenience, good patient compliance and low medicine production costs. In order for a drug to be absorbed into the systemic circulation following oral administration, the drug must be dissolved in the gastric fluids. For hydrophobic drugs, the dissolution process acts as the rate controlling step and, which determines the rate and degree of absorption. Thus, one of the major challenges to drug development today is poor solubility, as an estimated 40% of all newly developed drugs are poorly soluble or insoluble in water.1 In addition, up to 50% of orally administered drug compounds suffer from formulation problems related to their low solubility and high lipophilicity. Bioavailability of poorly water-soluble hydrophobic drugs [class II in biopharmaceutics classification system] is limited by their solubility and dissolution rate. The dissolution rate of these drugs can be improved by decreasing particle size, decreasing crystallinity, and/or increasing the surface area. Several studies have been carried out to increase the dissolution rate of drugs by decreasing the particle size. However, the fine drug particles have high tendency to agglomerate due to sander walls attraction or hydrophobicity, which both result in a decrease in surface area over time.2-4 Solid dispersion techniques have been used to enhance the dissolution and oral bioavailability of many poorly soluble drugs. The enhancement of oral bioavailability of such poorly water-soluble drugs remains one of the most challenging aspects of drug development. The development of solid dispersions as a practically viable method to enhance bioavailability of poorly water-soluble drugs overcame the limitations of previous approaches such as salt formation, solubilization by co solvent drugs in solid dispersion need not necessarily exist in the micronized state. A fraction of the drug might molecularly disperse in the matrix, thereby forming a solid dispersion.6, 7 When the solid dispersion is exposed to aqueous media, the carrier dissolves and the drug releases as fine colloidal particles. The resulting enhanced surface area produces higher dissolution rate and bioavailability of poorly water-soluble drugs. In addition, in solid dispersions, a portion of drug dissolves immediately to saturate the gastrointestinal tract fluid, and excess drug precipitates as fine colloidal particles or oily globules of submicron size.
Definitions of solid dispersion
The term solid dispersion refers to the dispersion of one or more active ingredients in an inert carrier or matrix at solid state prepared by the melting (fusion), solvent or melting solvent method.
Solid Dispersions
The term solid dispersion refers to a group of solid products consisting of at least two different components, generally a hydrophilic matrix and a hydrophobic drug. The matrix can be either crystalline or amorphous. The drug can be dispersed molecularly, in amorphous particles (clusters) or in crystalline particles.8, 9 Therefore, based on their molecular arrangement, six different types of solid dispersions can be distinguished. They are described in section 1.1.3. Moreover, certain combinations can be encountered, i.e. in the same sample; some molecules are present in clusters while some are molecularly dispersed.
Literature Review
1.Amol S. Deshmukh 2024 et al
Poor solubility is a common challenge in pharmaceuticals, hindering oral bioavailability. High throughput screening has led to an increase in poorly soluble drug candidates. Enhancing solubility and dissolution rates is crucial for drug development. Various methods, including soliddispersion, aim to improve solubility. A solid dispersion formulation process involves dispersing one or more active chemicals in a solidstate withinn an inert carrier or matrix. It can be made using solvent, melting, or melting-solvent procedures, among other techniques. By increasing the surface area and dispersibility of poorly soluble pharmaceuticals, this method improves their solubility and rate of dissolution, ultimately leading to an improvement in bioavailability
2.M. Sai Vishnu 2023 etal
The present work was aimed to enhancement of solubility of Nifedipine by using solid dispersion technique by kneading technique. Nifedipine is a class II drug and hence it is formulated by using solid dispersion technique to increase solubility of drug with the aid of polyvinyl pyrrolidine. Solid dispersion was prepared by combining of drug and carrier. The physical mixtures were prepared by solvent evaporation technique accordingly with the composition given in table. The ingredients like drug and carrier were accurately weighed (in different ratios 1:1, 1:1.5, 1:2, 1:2.5 1:3, 1:3.5, 1:4) and mixed in porcelain dish with a stirrer for 10 min to get uniform mix by using methanol. The above substance was dried. These are sieved to obtain granules by using sieve no.20. The blended powder was filled into capsules. The observed results reveals that carrier have significant effect on drug release up to 1 hr after successful development of solid dispersion containing Nifedipine and polyvinyl pyrrolidine carrier in different ratios (1:1, 1:1.5, 1:2, 1:2.5 1:3, 1:3.5, 1:4).
3.Sukannika Tubtimsri 2021 etal
Ternary solid solutions composed of nifedipine (NDP), amino methacrylate copolymer (AMCP), and polysorbate (PS) 20, 60, or 65 were prepared using a solvent evaporation method. The dissolution profiles of NDP were used to study the effect of the addition of polysorbate based on hydrophilic properties. A solid solution of NDP and AMCP was recently developed; however, the dissolution of NDP was <70%. In the present study, polysorbate was added to improve the dissolution of the drug by altering its hydrophilicity. The suitable formulation contained NDP and AMCP at a ratio of 1:4 and polysorbate at a concentration of 0.1%, 0.3%, or 0.6%. Differential scanning calorimetry and powder X-ray diffraction were used to examine the solid solutions. No peak representing crystalline NDP was observed in any solid solution samples, suggesting that the drug was molecularly dispersed in AMCP. The NDP dissolution from NDP powder and solid solution without PS were 16.82% and 58.19%, respectively. The highest dissolution of NDP of approximately 95.25% was noted at 120 min for the formulation containing 0.6% PS20. Linear correlations were observed between the surface free energy and percentages of dissolved NDP (R2 = 0.7115–0.9315). Cellular uptake across Caco-2 was selected to determine the drug permeability. The percentages of cellular uptake from the NDP powder, solid solution without and with PS20 were 0.25%, 3.60%, and 7.27%, respectively.
4.Nemanja Todorovi? 2024 etal
Supercritical fluid technology (SFT) is an insufficiently investigated approach for the production of solid dispersions; it is environmentally acceptable and has a high potential for application in the pharmaceutical industry. The aim of this work was to formulate and characterize nifedipine solid dispersions (SDs) produced by the SFT and compare the results with ones obtained by the classical solvent based kneading method. The following in vitro tests were conducted: assay and yield, solvent residues, solid state characterization (FTIR, DSC, XRD), flowability, hygroscopicity, solubility, dissolution and stability. Additionally, bioavailability was examined on an animal model (Wistar rats).
5.Minal Rajebahadur 2006 etal
The objective of our study was to find mechanisms responsible for solubility enhancement of nifedipine in solid dispersions of vitamin E TPGS and/or solutol HS-15. Solid dispersions of nifedipine with selected polymers such as vitamin E TPGS, solutol HS-15, PEG1000, and lipocol C-10 of varying drug/polymer ratios were prepared by a fusion method. The solubility enhancement was found to be in the order of vitamin E TPGS > solutol HS-15 > lipocol C-10 > PEG1000. Lipocol C-10, with a similar hydrophilic-lipophilic value as vitamin E TPGS, showed a comparable retained solubility enhancement during saturation solubility studies but had lower dissolution profile. Overall, vitamin E TPGS showed the best solubility and dissolution performance, while solutol HS-15 and lipocol C-10 demonstrated moderate solubility enhancements. Solid dispersions of vitamin E TPGS as prepared by microfluidization technique initially showed slightly higher solubility compared with samples prepared by fusion method, but eventually it became the same as the study progressed. However, solid dispersion of solutol HS-15 as prepared by microfluidization demonstrated a significant, sustained increased in solubility over its sample when prepared by fusion method.
6.Miss. Sneha V. Pawar1 2025 etal
The goal of enhancing Nifedipine's solubility is to improve its dissolution rate, particularly for BCS Class-II drugs. Researchers have successfully increased. Nifedipine's solubility using combinations of ethanol, sorbitol, propylene glycol, and PEG-polymers through co-solvency. Additionally, solubilization with polyoxyethylene sorbitol and hydrotropy using sodium salicylate, sodium benzoate, and sodium glycerate have also been employed.
2.1 Drug Profile: Nifedipine
2.2.1. Introduction68
Nifedipine is a calcium ion influx inhibitor (slow-channel blocker or calcium ion antagonist) which inhibits the transmembrane influx of calcium ions into vascular smooth muscle and cardiac muscle. The contractile processes of vascular smooth muscle and cardiac muscle are dependent upon the movement of extracellular calcium ions into these cells through specific ion channels. Nifedipine selectively inhibits calcium ion influx across the cell membrane of vascular smooth muscle and cardiac muscle without altering serum calcium concentrations.
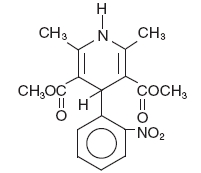
Chemical name: 3, 5-pyridinedicarboxylic acid, 1,4-dihydro-2,6-dimethyl-4-(2-nitrophenyl)-dimethyl ester
Non-proprietary name: Nifedipine
Proprietary name: Adalat, Procardia, Afeditab, Nifediac
CAS no: 21829-25-4
Empirical formula: C17H18N2O6
Molecular weight: 346.34
Biopharmaceutical classification system: Class-II (High Permeability & Low Solubility).
Structural formula:
2.2 Physicochemical Profile:
Description: Nifedipine is a yellow crystalline substance
Melting point: 171-175°c
Boiling point: 475°c
Solubility: Practically insoluble in water but soluble in ethanol.
Partition coefficient: Log P=3.17
Storage conditions: Sensitive to light
2.2.3. Pharmaceutical Profile:
Dosage forms marketed:
Extended-release tablets in the strengths of 30mg/60mg and soft gelatine capsules in the strengths of 10mg /20mg.
Stability and storage: Stable at room temperature, but to be protected from light.
2.2.5. Pharmacological Profile
Mechanism of action
The mechanism by which nifedipine reduces arterial blood pressure involves peripheral arterial vasodilatation and, consequently, a reduction in peripheral vascular resistance. The increased peripheral vascular resistance, an underlying cause of hypertension, results from an increase in active tension in the vascular smooth muscle. Studies have demonstrated that the increase in active tension reflects an increase in cytosolic free calcium. Nifedipine is a peripheral arterial vasodilator which acts directly on vascular smooth muscle. The binding of nifedipine to voltage-dependent and possibly receptor-operated channels in vascular smooth muscle results in an inhibition of calcium influx through these channels. Stores of intracellular calcium in vascular smooth muscle are limited and thus dependent upon the influx of extracellular calcium for contraction to occur. The reduction in calcium influx by nifedipine causes arterial vasodilation and decreased peripheral vascular resistance which results in reduced arterial blood pressure. The precise means by which this inhibition relieves angina has not been fully determined, but includes at least the following two mechanisms:
Nifedipine dilates the main coronary arteries and coronary arterioles, both in normal and ischemic regions, and is a potent inhibitor of coronary artery spasm, whether spontaneous or ergonovine-induced. This property increases myocardial oxygen delivery in patients with coronary artery spasm, and is responsible for the effectiveness of nifedipine in vasospastic (Prinzmetal's or variant) angina. Whether this effect plays any role in classical angina is not clear, but studies of exercise tolerance have not shown an increase in the maximum exercise rate-pressure product, a widely accepted measure of oxygen utilization. This suggests that, in general, relief of spasm or dilation of coronary arteries is not an important factor in classical angina.
Nifedipine regularly reduces arterial pressure at rest and at a given level of exercise by dilating peripheral arterioles and reducing the total peripheral resistance (afterload) against which the heart works. This unloading of the heart reduces myocardial energy consumption and oxygen requirements and probably accounts for the effectiveness of nifedipine in chronic stable angina.
Pharmacodynamics
A variety of clinical studies have demonstrated that like other slow-channel blockers, nifedipine exerts a negative inotropic effect on isolated myocardial tissue. This is rarely, if ever, seen in intact animals or man, probably because of reflex responses to its vasodilating effects. In man, nifedipine causes decreased peripheral vascular resistance and a fall in systolic and diastolic pressure, usually modest (5–10 mm Hg systolic), but sometimes larger. There is usually a small increase in heart rate, a reflex response to vasodilation. Measurements of cardiac function in patients with normal ventricular function have generally found a small increase in cardiac index without major effects on ejection fraction, left ventricular end diastolic pressure (LVEDP) or volume (LVEDV). In patients with impaired ventricular function, most acute studies have shown some increase in ejection fraction and reduction in left ventricular filling pressure.
Pharmacokinetics
Nifedipine, when given in capsules of 10 mg gets rapidly and fully absorbed after oral administration. The drug is detectable in serum 10 minutes after oral administration, and peak blood levels occur in approximately 30 minutes. Bioavailability is proportional to dose from 10 to 30 mg; half-life does not change significantly with dose. It is highly bound by serum proteins (92-98%). Nifedipine is extensively converted to inactive metabolites and approximately 80 percent of nifedipine and metabolites are eliminated via the kidneys. The half-life of nifedipine in plasma is approximately two hours. Since hepatic biotransformation is the predominant route for the disposition of nifedipine, the pharmacokinetics may be altered in patients with chronic liver disease. Patients with hepatic impairment (liver cirrhosis) have a longer disposition half-life and higher bioavailability of nifedipine than healthy volunteers. The degree of serum protein binding of nifedipine is high (92-98%). Protein binding may be greatly reduced in patients with renal or hepatic impairment. In healthy subjects, the elimination half-life of a sustained release nifedipine formulation was longer in elderly subjects (6.7 h) compared to young subjects (3.8 h) following oral administration. A decreased clearance was also observed in the elderly (348 ml/min) following intravenous administration. The drug is excreted through urine (60% to 80% as inactive metabolites) and feces. Co-administration of nifedipine with grapefruit juice resulted in approximately a 2-fold increase in nifedipine AUC and Cmax with no change in half-life. The increased plasma concentrations are most likely due to inhibition of CYP 3A4 related first-pass metabolism.
Indications and uses
Nifedipine extended-release tablets are indicated for the management of vasospastic angina by any of the following criteria: 1) classical pattern of angina at rest accompanied by ST segment elevation, 2) angina or coronary artery spasm provoked by ergonovine, or 3) angiographically demonstrated coronary artery spasm. In those patients who have had angiography, the presence of significant fixed obstructive disease is not incompatible with the diagnosis of vasospastic angina, provided that the above criteria are satisfied. Nifedipine extended-release may also be used where the clinical presentation suggests a possible vasospastic component but where vasospasm has not been. e.g., where pain has a variable threshold on exertion or in unstable angina where electrocardiographic findings are compatible with intermittent vasospasm, or when angina is refractory to nitrates and/or adequate doses of beta blockers.
Nifedipine extended-release tablets are indicated for the management of chronic stable angina (effort-associated angina) without evidence of vasospasm in patients who remain symptomatic despite adequate doses of beta blockers and/or organic nitrates or who cannot tolerate those agents. In chronic stable angina (effort-associated angina) nifedipine has been effective in controlled trials of up to eight weeks duration in reducing angina frequency and increasing exercise tolerance, but conformation of sustained effectiveness and evaluation of long-term safety in these patients is incomplete. Controlled studies in small numbers of patients suggest concomitant use of nifedipine and beta-blocking agents may be beneficial in patients with chronic stable angina, but available information is not sufficient to predict with confidence the effects of concurrent treatment, especially in patients with compromised left ventricular function or cardiac conduction abnormalities. When introducing such concomitant therapy, care must be taken to monitor blood pressure closely since severe hypotension can occur from the combined effects of the drugs.
Nifedipine extended-release tablets are indicated for the treatment of hypertension. They may be used alone or in combination with other antihypertensive agents.
Drug interactions
Calcium Channel Blockers
Diltiazem: Pre-treatment of healthy volunteers with 30 mg or 90 mg t.i.d. diltiazem p.o. increased the AUC of nifedipine after a single dose of 20 mg nifedipine by factors of 2.2 and 3.1, respectively.
ACE Inhibitors
Benazepril: A hypotensive effect was only seen after co-administration of the two drugs. The tachycardic effect of nifedipine was attenuated in the presence of benazepril.
Digoxin: The simultaneous administration of nifedipine and digoxin may lead to reduced clearance resulting in an increase in plasma concentrations of digoxin.Antithrombotic
Platelet Aggregation Inhibitors
Clopidogrel: No clinically significant pharmacodynamic interactions were observed when clopidogrel was co-administered with nifedipine.
Diuretics, PDE5 inhibitors, alpha-methyldopa:
Nifedipine may increase the blood pressure lowering effect of these concomitantly administered agents.
Antifungal Drugs
Ketoconazole, itraconazole and fluconazole are CYP3A inhibitors and can inhibit the metabolism of nifedipine and increase the exposure to nifedipine during concomitant therapy. Blood pressure should be monitored and a dose reduction of nifedipine considered.
CNS Drugs
Fluoxetine, a CYP3A inhibitor, can inhibit the metabolism of nifedipine and increase the exposure to nifedipine during concomitant therapy. Blood pressure should be monitored and a reduction of the dose of nifedipine considered. Valproic acid may increase the exposure to nifedipine during concomitant therapy. Blood pressure should be monitored and a dose reduction of nifedipine considered.
Immunosuppressive Drugs
Tacrolimus: Tacrolimus has been shown to be metabolized via the CYP3A system. Nifedipine has been shown to inhibit the metabolism of tacrolimus in vitro.
Glucose Lowering Drugs
Rosiglitazone: Co-administration of rosiglitazone (4 mg b.i.d.) was shown to have no clinically relevant effect on the pharmacokinetics of nifedipine.
Miglitol: No effect of miglitol was observed on the pharmacokinetics and pharmacodynamics of nifedipine.
2.2 Excipients Profile:
2.2.1 Croscarmellose sodium (CCS)
1. Nonproprietary names: BP: Croscarmellose sodium, PhEur: Croscarmellose sodium, USP-IR: Croscarmellose sodium
2. Synonyms: Ac-Di-Sol; cross linked carboxymethylcellulose
sodium; Explocel; modified cellulose gum; Nymcel ZSX; Pharmacel XL; Primellose; Solutab; Vivasol.
3. Chemical name and CAS registry number:
Cellulose, carboxymethyl ether, sodium salt, cross linked [74811-65-7]
4. Functional category: Tablet and capsule disintegrants
5. Structural formula:
Fig: 2.3 Structure of Croscarmellose sodium
6. Applications in pharmaceutical formulation or technology:
Croscarmellose sodium is used in oral pharmaceutical formulations as a disintegrant for capsules, (10-25%) tablets (0.5%-5%). In tablet formulations, croscarmellose sodium may be used in both direct-compression and wet-granulation processes. When used in wet granulations, as intra and extra granular portions wicking and swelling ability of the disintegrant is best utilized. Croscarmellose sodium at concentrations up to 5% w/w may be used as a tablet disintegrant, although normally 2% w/w is used in tablets prepared by direct compression and 3% w/w in tablets prepared by a wet-granulation process.
7. Description: Croscarmellose sodium occurs as an odorless, white or grayish white powder.
8. Solubility: Insoluble in water, although croscarmellose sodium rapidly swells to 4–8 times its original volume on contact with water. Practically insoluble in acetone, ethanol and toluene.
9. Safety: Croscarmellose sodium is mainly used as a disintegrant in oral pharmaceutical formulations and is generally regarded as an essentially nontoxic and nonirritant material. However, oral consumption of large amounts of croscarmellose sodium may have a laxative effect, although the quantities used in solid dosage formulations are unlikely to cause such problems.
2.2.2 Sodium Starch Glycolate (SSG)
Fig: 2.4 Structure of Sodium starch glycolate
2.2.3 Crospovidone
1. Nonproprietary names: BP: crospovidone, PhEur: crospovidone, USP-IR: crospovidone
2. Synonyms: Crospovidonum; Crospopharm; cross linked povidone; E1202; Kollidon CL; Kollidon CL-M; Polyplasdone XL; Polyplasdone XL-10; polyvinylpolypyrrolidone; PVPP; 1-vinyl-2-pyrrolidinone homopolymer.
3 Chemical name and CAS registry number: 1-Ethenyl-2-pyrrolidinone homopolymer [9003-39-8]
4. EmpIRical formula and molecular weight: (C6H9NO) n >1 000 000
5. Structural formula:
6. Functional category: Tablet disintegrant.
7. Applications in pharmaceutical formulation or technology:
Crospovidone is a water-insoluble tablet disintegrant and dissolution agent used at 2–5% concentration in tablets prepared by direct compression or wet- and dry-granulation methods. It rapidly exhibits high capillary activity and pronounced hydration capacity, with little tendency to form gels. Crospovidone can also be used as a solubility enhancer. With the technique of co-evaporation, crospovidone can be used to enhance the solubility of poorly soluble drugs. The drug is adsorbed on to crospovidone in the presence of a suitable solvent and the solvent is then evaporated. This technique results in faster dissolution rate.
8. Description: Crospovidone is a white to creamy-white, finely divided, free flowing, practically tasteless, odorless or nearly odorless, hygroscopic powder.
9. Solubility: Practically insoluble in water and most common organic solvents.
10. Safety: Crospovidone is used in oral pharmaceutical formulations and is generally regarded as a nontoxic and nonirritant material. Short term animal toxicity studies have shown no adverse effects associated with crospovidone. LD50 (mouse, IP): 12 g/kg.
2.2.4 Kyron T-314

Use level: 0.4% to 2% of average weight of tablet on lubrication stage.
Disintegrating agent: Kyron T314 has a very high swelling tendency on hydration either in contact with water or G.I. fluids causing fast disintegration without the formation of lumps and thus acts as an effective tablet disintegrant.
Toxicity: Kyron T-314 is high molecular weight polymer, so doesn't get absorbed by body tissues & is safe for human consumption. It has no physiological action at recommended dosage & it is non-toxic.
2.3 Research Envisaged
2.4 Aim & Objectives:
Aim: The main aim of this project is to enhance the solubility of Nifedipine by solid dispersion technique
Objectives:
• To prepare the solid dispersion of Nifedipine by solvent evaporation method using different carriers at different ratios.
• To evaluate the above prepared solid dispersion by FTIR, SEM and in vitro dissolution studies.
• Selection of optimized solid dispersion by using in vitro dissolution results.
• Preparation of oral dispersible tablets of Nifedipine prepared with optimized surface solid dispersion.
• Evaluation of oral dispersible tablets of Nifedipine for hardness, weight variation, friability, disintegration time and in vitro dissolution.
2.5 Plan of Work
Analytical method development.
Experimental Methodology
MATERIALS & METHODS
Table no 3.1: Equipment’s used for preparation and analysis
|
S. No |
Equipment |
Name |
|
1 |
Dissolution test apparatus –USP Type II |
Electrolab |
|
2 |
Disintegration test apparatus |
Electrolab |
|
3 |
Digital weighing machine |
Contech instruments limited |
|
4 |
Friability tester |
Electrolab |
|
5 |
IR spectrophotometer |
Shimadzu 8400S |
|
6 |
Monsanto hardness tester |
Monsanto |
|
7 |
Tablet punching machi |
Rimek |
|
8 |
Hot aIR oven |
OS world |
|
9 |
UV-Vis Double beam Spectrophotometer |
Schimadzu |
Table No 3.2: List of Materials Used
|
S. No |
Material Name
|
Manufacturer |
|
1 |
Nifedipine |
Dr. Reddy’s laboratories, Hyderabad. |
|
2 |
Croscarmellose sodium |
FMC BioPolymer |
|
3 |
Sodium starch glycolate |
DMV International |
|
4 |
Cyclodextrin |
DMV international |
|
5 |
Kyron-T-314 |
Corel pharma chem. |
|
6 |
Crospovidone |
BASF |
3.1 Analytical Method Development for Nifedipine
Standard curve of nifedipine in 6.8 pH phosphate buffer solution containing 0.5% SLS media was performed to quantify the samples. All solutions were freshly prepared before use.
Preparation of standard solution of nifedipine
Accurately weighed 100 mg of nifedipine was placed in a 100 ml volumetric flask. 15 ml of methanol was added to dissolve drug. The volume was made up to 100 ml using 6.8 pH phosphate buffer solution containing 0.5% SLS to give 1000 mcg/ml solution (stock solution – I). A 10 ml aliquot was taken in a 100 ml volumetric flask and diluted with 6.8 pH phosphate buffer solution containing 0.5% SLS to 100 ml to get 100 mcg/ml (stock solution -II). From stock solution -II, a 10 ml of aliquot was made up to 100 ml with 6.8 pH phosphate buffer solution containing 0.5% SLS to get 10 mcg/ml (stock solution-III).
Determination of absorption maxima (λmax) for nifedipine65
A 10 mcg/ml standard solution of nifedipine was scanned on a double beam spectrophotometer against respective media blanks. An absorption maximum (λmax) of 238 nm was obtained for all solutions and standard curve was prepared.
Preparation of standard curve for nifedipine
Standard curves for nifedipine were obtained in 6.8 pH phosphate buffer solution containing 0.5% SLS. Aliquots of 2, 4, 6, 8, and 10 ml of nifedipine standard solution of 10 mcg/ml (stock solution-III) were taken and diluted to obtain concentrations from 2,4,6,8 and10 mcg/ml respectively with buffer media. Similarly, 20 ml aliquot of nifedipine standard solution of 100 mcg/ml (stock solution-II) was taken and diluted with buffer medium to obtain 20 mcg/ml respectively. The absorbances of solutions were determined at 238 nm against respective media as blanks. The experiment was repeated six times for the medium and a calibration curve was determined from the mean value.
3.2 Preparation and characterization of physical mixtures of Nifedipine prepared by blending method
The physical mixtures of Nifedipine with different carriers were prepared by blending method. The physical mixtures were prepared at various drug to carrier ratios with all the carriers in the increasing order of carrier concentrations. The prepared mixtures were characterized by in vitro dissolution studies for the improvement in release compared to that of the pure drug. The mixtures which showed the improvement in release considerably were selected and optimized for further studies.
Preparation, characterization and optimization of solid dispersions of Nifedipine prepared by solvent evaporation method
The solid dispersions of Nifedipine were prepared with various carriers by solvent evaporation method. The solid dispersions were prepared at various drug to carrier ratios with all the carriers in the increasing order of carrier concentration. The prepared dispersions were characterized by in vitro dissolution studies for the improvement in release compared to that of the pure drug and physical mixtures. The dispersions which showed the improvement in release considerably were selected and optimized for further studies.
Preparation of physical mixtures (PM) of Nifedipine by blending method.
The physical mixtures of Nifedipine were prepared with the blends of drug and carriers in different ratios with increasing concentration of the carriers. These blends were prepared by mixing drug and the required amount of carrier in a mortar and pestle for 20 minutes. The ratios and weights of the ingredients taken are shown in the Table nos below. Blends were mixed thoroughly and were passed through 60 mesh. These mixtures were then stored in a desiccator for further characterization.
Table no 3.3: Formulas of PMs of Nifedipine and croscarmellose sodium (CCS)
|
Formula code |
Ratio of the mixture of Nifedipine and CCS (D:C) |
Weight of the drug (mg) |
Weight of the carrier (mg) |
|
PMC-1 |
1:1 |
150 |
150 |
|
PMC-2 |
1:3 |
150 |
450 |
|
PMC-3 |
1:5 |
150 |
750 |
NOTE: D-drug (Nifedipine), C-carrier (croscarmellose sodium)
Table no 3.4: Formulas of PMs of Nifedipine and sodium starch glycolate (SSG).
|
Formula code |
Ratio of the mixture of Nifedipine and SSG (D:C) |
Weight of the drug (mg) |
Weight of the carrier (mg) |
|
PMS-1 |
1:1 |
150 |
150 |
|
PMS-2 |
1:3 |
150 |
450 |
|
PMS-3 |
1:5 |
150 |
750 |
NOTE: D-drug (Nifedipine), C-carrier (sodium starch glycolate)
Table no 3.5: Formulas of PMs of Nifedipine and β- cyclodextrin.
|
Formula code |
Ratio of the mixture of Nifedipine and β- cyclodextrin (D:C) |
Weight of the drug (mg) |
Weight of the carrier (mg) |
|
PMS-1 |
1:1 |
150 |
150 |
|
PMS-2 |
1:3 |
150 |
450 |
|
PMS-3 |
1:5 |
150 |
750 |
NOTE: D-drug (Nifedipine), C-carrier (β- cyclodextrin)
Characterization of the prepared physical mixtures
The PMs prepared were analyzed for assay and invitro release of drug by dissolution studies.
Assay of the physical mixtures
Assay of the prepared PMs were determined in 6.8 pH phosphate buffer solution. Accurately weighed amounts of PMs equivalent to 150 mg of drug were taken in a 100 ml volumetric flask, 20 ml ethanol was added and shaken for 20 min to dissolve the drug. The volume was made to 100 ml with buffer medium separately. Dispersions were filtered and 1 ml aliquot of the above solutions were taken and diluted to 10 ml with buffer medium respectively. The concentration of the resultant solution was 10 µg/ml. The absorbances of these solutions were determined at 238 nm against the blank. The percentage assay was calculated from the standard curve.
In vitro dissolution study of the physical mixtures
The prepared PMs were accurately weighed equivalent to 150 mg of the drug. These mixtures were analyzed for drug release in 900 ml of the dissolution media by powder dispersion technique. The dissolution media in which the tests were performed was 6.8 pH phosphate buffer solution. The samples were automatically withdrawn at time intervals 10 min, 20 min, 30 min, 40 min, 50 min and 60 min. Filters were used to avoid the solid particles from being withdrawn which might interfere in analysis. The samples were analyzed spectrophotometrically at the maximum wavelength (λmax) of the drug which is 238nm against the blank media. Dissolution of each sample was performed 3 times (n=3) and mean of all determinations was used to calculate the drug release profile. The apparatus used was USP Type II with 50 rpm.
In vitro dissolution study of the plain drug
The standard dose of Nifedipine i.e., 150 mg was accurately weighed. These samples were analyzed for the drug release in the same manner as the PMs by powder dispersion technique. The release rate of the pure drug from the samples was compared to the release rate of the drug from prepared physical mixtures. The mixtures which improved the drug release rate considerably compared to that of the pure drug were selected for the further work
Data treatment of dissolution studies:
Dissolution profiles of percentage drug release vs. time were obtained. Amount of drug released at 60 minutes was calculated.
Preparation of solid dispersion of Nifedipine by Solvent evaporation method:
Nifedipine and each of surface-active carriers (CCS, SSG and Cyclodextrin) were weighed accurately in various ratios (1:1, 1:3 and 1:5) and transferred to China dish containing sufficient quantity of ethanol to dissolve. Ethanol was evaporated on heating mantle at 400C. The resulting solid dispersions were stored for 24 hrs in desiccator to congeal. The mass obtained was crushed, pulverized. Finally, dispersions were passed through sieve number #40 and were stored in air tight containers till further use.
Table 3.6 Formulas of SSDs of Nifedipine and croscarmellose sodium
|
Formula code |
Ratio of the mixture of Nifedipine and CCS (D:C) |
Weight of the drug (mg) |
Weight of the carrier (mg) |
|
SDC-1 |
1:1 |
150 |
150 |
|
SDC-2 |
1:3 |
150 |
450 |
|
SDC-3 |
1:5 |
150 |
750 |
Note: D-drug (Nifedipine), C-carrier (croscarmellose sodium)
Table 3.7 Formulas of SDs of Nifedipine and sodium starch glycolate.
|
Formula code |
Ratio of the mixture of Nifedipine and SSG (D:C) |
Weight of the drug (mg) |
Weight of the carrier (mg) |
|
SDS-1 |
1:1 |
150 |
150 |
|
SDS-2 |
1:3 |
150 |
450 |
|
SDS-3 |
1:5 |
150 |
750 |
Note: D-drug (Nifedipine), C-carrier (sodium starch glycolate)
Table 3.8 Formulas of SSDs of Nifedipine and β- cyclodextrin.
|
Formula code |
Ratio of the mixture of β- cyclodextrin and SSG (D:C) |
Weight of the drug (mg) |
Weight of the carrier (mg) |
|
SCD-1 |
1:1 |
150 |
150 |
|
SCD-2 |
1:3 |
150 |
450 |
|
SCD-3 |
1:5 |
150 |
750 |
Note: D-drug (Nifedipine), C-carrier (β- cyclodextrin)
3.3 Characterization of the prepared solid dispersions of Nifedipine
The assay of all the dispersions was performed by UV spectroscopy. The prepared dispersions were analyzed for the physical properties by spectral analysis using FTIR, for release rate by in vitro dissolution study.
In-vitro dissolution study of the solid dispersions
In-vitro dissolution study of the solid dispersions of Nifedipine by using different carriers were performed in the same manner as that of the PMs prepared by blending method. The release rate of the solid dispersions was compared with that of pure drug and PMs to check for the improvement of the dissolution of surface solid dispersions. Results were checked for reproducibility. The improvement in dissolution was more with formula SCD-3, which was optimized and further characterized.
Spectral analysis by FTIR
The spectrum analysis of the prepared surface solid dispersion of Nifedipine of SCD-3 was studied by FTIR. FTIR spectra were recorded by preparing potassium bromide (KBr) disks using a Shimadzu Corporation (Koyto, Japan) facility (model - 8400S). Potassium bromide (KBr) disks were prepared by mixing few mg of sample with potassium bromide by compacting in a hydrostatic press under vacuum at 6-8 tons pressure. The resultant disc was mounted in a suitable no holder in IR spectrophotometer and the IR spectrum was recorded from 4000 cm-1 to 500 cm-1 in a scan time of 12 minutes. The resultant spectrum was compared for any spectral changes. They were observed for the presence of characteristic peaks for the respective functional group in the compound. The pure drug and β- cyclodextrin were analyzed by FTIR in same manner and were observed for the presence of respective peaks.
Surface morphology by SEM
The surface morphology of Nifedipine SSD on β- Cyclodextrin (1:5) and PM were observed by using an analytical scanning electron microscope (JSM- 6360A, JEOL, Tokyo, Japan). The samples were lightly sprinkled on a double-sided adhesive tape stuck to an aluminum stub. The stubs were then coated with platinum to a thickness of about 10 Å under an argon atmosphere using a gold-sputter module in a high-vacuum evaporator. Afterwards, the stubs containing the coated samples were placed in the scanning electron microscope chamber.
3.4 Preparation and Evaluation of Oral Dispersible Tablets of Nifedipine
Oral dispersible tablets were formulated with the optimized surface solid dispersion of Nifedipine and were evaluated.
Preparation of Oral Dispersible tablets with optimized surface solid dispersion of Nifedipine: 67
Oral dispersible tablets were prepared using optimized surface solid dispersion of Nifedipine SCD-3 with different disintegrants and without disintegrants (NFS) as per the formula given in Table no.3.9. The SSDs were thoroughly mixed with microcrystalline cellulose, disintegrants like Kyron T-314 (NFK) and crospovidone (NFCP) for 10 minutes. Finally, it is mixed with magnesium stearate and talc for 5 minutes. This mixture was then compressed into Tablets on a rotary Tablet punching machine using 8 mm punch.
Table no 3.9: Formulation for surface solid dispersion of Nifedipine (SCD-3) tablets
|
Ingredients (mg) |
NFS |
NFK
|
NFCP |
|
Irbesartan SCD-3(equivalent to 10 mg of irbesartan) |
109 |
109 |
109 |
|
SMCC |
30 |
30 |
30 |
|
MCC |
34 |
30 |
30 |
|
Mannitol |
20 |
20 |
20 |
|
Sodium saccharin |
4 |
4 |
4 |
|
Magnesium stearate |
2 |
2 |
2 |
|
Talc |
1 |
1 |
1 |
|
Kyron T-314 |
|
4 |
|
|
Crospovidone |
|
|
4 |
|
Total |
200 mg |
200 mg |
200 mg |
3.5 Evaluation of Oral dispersible tablets of Nifedipine surface solid dispersion:
Tablets prepared were evaluated for parameters as described below.
Weight variation: Twenty tablets were selected randomly from each batch and weighed. The weight variation test was performed as per Indian pharmacopoeia.
Hardness: Hardness of tablet was determined using a Monsanto tablet hardness tester.
Friability: Friability of the tablets was determined using a Roche friabilator. Accurately weighed six tablets were placed in the friabilator and rotated at 25 rpm for 4 min.
Percentage friability was calculated using the following equation.
Friability = ([ wO – w] /wO) ´ 100
Where; wO = weight of the Tablet at time zero before revolution.
w = weight of the Tablet after 100 revolutions.
Disintegration time: Disintegration times of tablets were determined, using water at 370C as medium as described in Indian pharmacopoeia.
Assay: Ten oral dispersible tablets of Nifedipine surface solid dispersion were crushed in a motor and pestle. Sample equivalent to 10 mg of Nifedipine was weighed accurately and analyzed as described in section 3.5.2 under “Assay”.
Dissolution studies of Tablets: In vitro dissolution studies of tablets were carried out using USP apparatus II paddle method. Nifedipine tablets were placed in 900 ml of the solution containing 6.8 pH phosphate buffer solution containing 0.5% SLS at 37+0.5°C and speed at 50 rpm. Samples were withdrawn and analyzed as described under “In-vitro dissolution studies” in section 3.5.2
RESULTS AND DISCUSSION
4.1 Analytical Method For Nifedipine:
λ max of nifedipine
The analytical method development of nifedipine was performed for the determination of its λmax and quantification of the dispersions before proceeding for the experiment. Nifedipine was scanned in 6.8 pH phosphate buffer containing 0.5% SLS to determine the λmax. An absorption maximum of 238 nm is obtained (fig.4.1) A standard graph of nifedipine in 6.8 pH phosphate buffer containing 0.5% SLS medium is prepared at this λmax
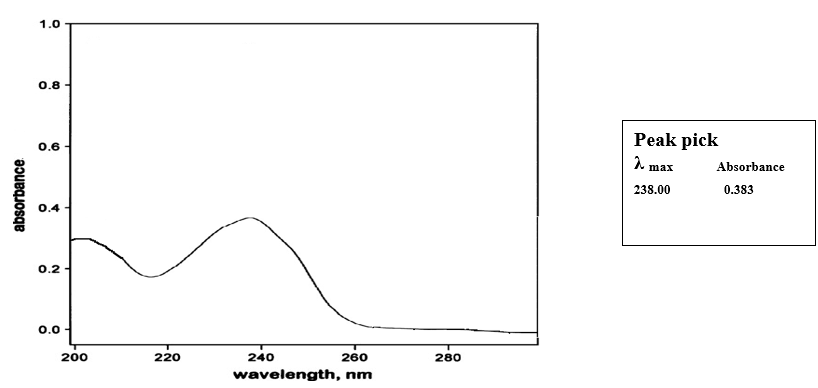
Fig no 4.1: λmax Scan of nifedipine
Preparation of standard graph:
Standard solutions in the range of 2 to 20 mcg/ml were prepared and absorption values were recorded at 238 nm against the blank. From this data, the standard curve of Nifedipine was obtained by plotting absorbance on Y-axis against concentration on X-axis.
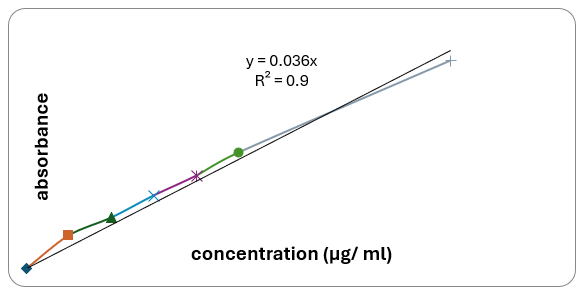
Fig no 4.1: Standard graph of Nifedipine in 6.8 pH phosphate buffer containing 0.5 % SLS
4.2 Selection Of Carriers
Carriers’ selection plays a crucial role in the preparation of physical mixtures. Carriers selected in the present work are croscarmellose sodium, sodium starch glycolate and β-cyclodextrin. The objective in the present study is to show that different carriers at different increasing ratios enhance the dissolution of the poorly soluble drugs.
4.3 Preparation and Characterization of Physical Mixtures of Nifedipine Prepared by Blending Method.
Preparation of PMs of Nifedipine by blending method.
The PMs of Nifedipine were prepared with the blends of drug and carriers in different ratios with increasing concentration of the carriers.
The PMs prepared above were analysed for assay and the improvement in dissolution rate of the drug Nifedipine using various carriers by in vitro dissolution studies.
Assay and uniformity content:
The PMs prepared complied with the requirements of assay and content uniformity. The results for assay and content uniformity were 98.25 ±4.2% and 97.74±5.4% respectively. These percentage drug values indicated that the drug content is uniform in the batch of PMs in all cases.
Drug release study by in-vitro drug dissolution:
The prepared PMs were evaluated for the drug release by invitro dissolution study. Dose equivalent to 150 mg is weighed. The apparatus used is USP Type II with 50 RPM. The media in which the study is done is 6.8 pH phosphate buffer. The volume of the media is 900 ml. Samples were withdrawn at the time intervals 10, 20, 30-, 40-, 50- and 60-min. Samples were detected at the λmax of 238 nm.
Table 4.1: Percentage release profiles of Nifedipine PMs with sodium starch glycolate as carrier in 6.8 pH phosphate buffer
|
Time(mins) |
Pure drug |
PMS-1 |
PMS-2 |
PMS-3 |
|
0 |
0 |
0 |
0 |
0 |
|
10 |
13.44 |
15.11 |
15.66 |
15.66 |
|
20 |
23.24 |
29.91 |
32.14 |
32.14 |
|
30 |
32.81 |
38.97 |
40.93 |
41.76 |
|
40 |
34.66 |
41.68 |
42.54 |
42.82 |
|
50 |
38.18 |
44.69 |
45.28 |
46.11 |
|
60 |
44.78 |
50.21 |
50.52 |
50.81 |
Note: PMS- physical mixture of sodium starch glycolate.
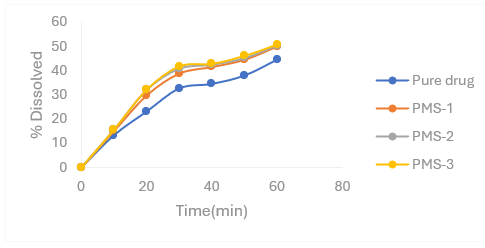
Figure 4.1: Percentage release profiles of Nifedipine PMs with sodium starch glycolate
Table 4.2 Percentage release profiles of Nifedipine PMs with croscarmellose sodium as carrier in 6.8 pH phosphate buffer
|
Time (mins) |
Pure drug |
PMC-1 |
PMC-2 |
PMC-3 |
|
0 |
0 |
0 |
0 |
0 |
|
10 |
13.44 |
15.11 |
16.22 |
17.88 |
|
20 |
23.24 |
30.47 |
31.86 |
32.98 |
|
30 |
32.81 |
39.53 |
41.48 |
43.17 |
|
40 |
34.66 |
41.97 |
43.38 |
45.07 |
|
50 |
38.18 |
45.25 |
46.40 |
48.65 |
|
60 |
44.78 |
50.78 |
52.48 |
53.64 |
Note: PMC- Physical Mixture of Croscarmellose Sodium.
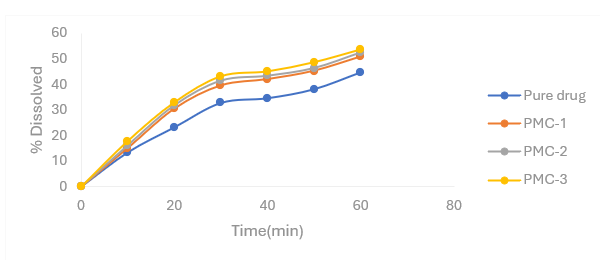
Figure 4.3: Percentage release profiles of Nifedipine PMs with croscarmellose sodium
Table 4.3 Percentage release profiles of Nifedipine PMs with β- cyclodextrin as carrier in 6.8 pH phosphate buffer
|
Time (mins) |
Pure drug |
PCD-1 |
PCD-2 |
PCD-3 |
|
0 |
0 |
0 |
0 |
0 |
|
10 |
13.44 |
16.22 |
17.33 |
18.72 |
|
20 |
23.24 |
31.86 |
32.42 |
33.82 |
|
30 |
32.81 |
40.93 |
42.05 |
43.45 |
|
40 |
34.66 |
43.38 |
44.50 |
45.92 |
|
50 |
38.18 |
46.67 |
47.25 |
49.50 |
|
60 |
44.78 |
53.04 |
53.89 |
56.16 |
Note: PCD- physical mixture of β- cyclodextrin
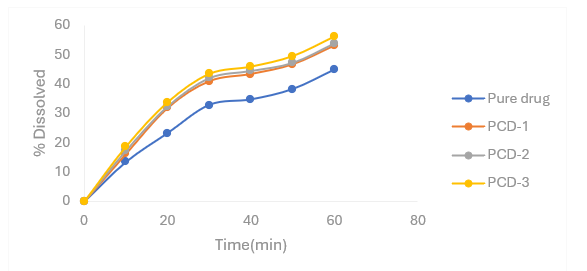
Figure 4.4: Percentage release profiles of Nifedipine PMs with β- cyclodextrin as carrier in 6.8 pH phosphate buffer
4.3 Preparation, Characterization and Optimization of Solid Dispersions of Nifedipine Prepared by Solvent Evaporation Method
Preparation of SDs of Nifedipine by solvent evaporation method
The SDs were prepared at various drug to carrier ratios with all the carriers in the increasing order of carrier concentration by solvent evaporation method. This method provides uniform distribution of drug than when compared to blending method in a fine form by solubilizing the drug in a suitable solvent and thereby depositing on a suitable carrier. The prepared dispersions were characterized by in vitro dissolution studies for the improvement in release compared to that of the pure drug and physical mixtures. The dispersions which showed the improvement in release considerably were selected and optimized for further studies.
Characterization of the prepared solid dispersions of Nifedipine
The assay of all dispersions was performed by UV spectrophotometry. The prepared dispersions were analyzed for the release rate behavior by in vitro dissolution.
Assay and uniformity content: Assay of the prepared different solid dispersions was performed and the prepared dispersions complied with the requirements of assay and content uniformity. The results for assay and content uniformity were 97.85 ±6.9% and 98.02±7.6% respectively. These drug content values indicated that the drug content was uniform in the batch of surface solid dispersions.
Drug release study by in-vitro drug dissolution: The prepared solid dispersions were evaluated for the drug release by in vitro dissolution study. Dose equivalent to 150 mg was weighed. The apparatus used was USP Type II with 50 RPM. The media in which the study was done was 6.8 pH phosphate buffer. The volume of the media was 900 ml. Samples were withdrawn at the time intervals 10, 20, 30-, 40-, 50- and 60-min. Samples were detected at the λmax of 244 nm.
Table 4.4 Percentage release profiles of Nifedipine SDs with croscarmellose sodium as carrier in 6.8 pH phosphate buffer
|
Time(mins) |
Pure drug |
SDC-1 |
SDC-2 |
SDC-3 |
|
0 |
0 |
0 |
0 |
0 |
|
10 |
13.44 |
60.11 |
61.77 |
62.88 |
|
20 |
23.24 |
71.00 |
72.12 |
74.07 |
|
30 |
32.81 |
76.39 |
78.07 |
80.07 |
|
40 |
34.66 |
80.70 |
82.67 |
83.52 |
|
50 |
38.18 |
84.2 |
85.90 |
86.61 |
|
60 |
44.78 |
86.88 |
88.31 |
88.08 |
Note: SDC- solid dispersion of croscarmellose sodium.
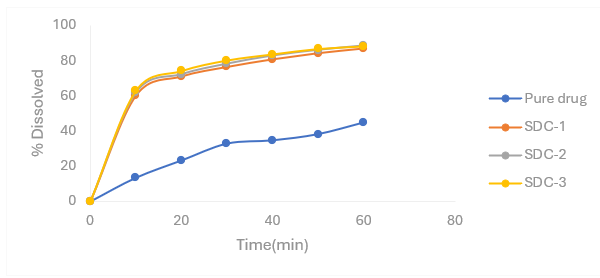
Figure 4.5: Percentage release profiles of Nifedipine SDs with croscarmellose sodium as carrier in6.8 pH phosphate buffer
Table 4.5 Percentage release profiles of Nifedipine SDs with sodium starch glycolate as carrier in 6.8 pH phosphate buffer
|
Time (mins) |
Pure drug |
SDS-1 |
SDS-2 |
SDS-3 |
|
0 |
0 |
0 |
0 |
0 |
|
10 |
13.44 |
54.83 |
55.94 |
56.77 |
|
20 |
23.24 |
62.63 |
64.58 |
66.53 |
|
30 |
32.81 |
68.53 |
70.22 |
72.18 |
|
40 |
34.66 |
74.19 |
75.88 |
77.30 |
|
50 |
38.18 |
76.26 |
78.52 |
79.67 |
|
60 |
44.78 |
80.01 |
82 |
82.88 |
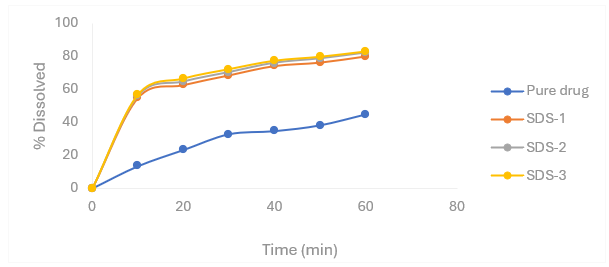
Figure 4.6 Percentage release profiles of Nifedipine SDs with sodium starch glycolate as carrier in 6.8 pH phosphate buffer
Table 4.6 Percentage release profiles of Nifedipine SDs with β- cyclodextrin as carrier in 6.8 pH phosphate buffer
|
Time(mins) |
Pure drug |
SCD-1 |
SCD-2 |
SCD-3 |
|
0 |
0 |
0 |
0 |
0 |
|
10 |
13.44 |
64.27 |
65.94 |
67.61 |
|
20 |
23.24 |
76.85 |
79.92 |
80.48 |
|
30 |
32.81 |
82.83 |
85.64 |
87.59 |
|
40 |
34.66 |
84.96 |
87.77 |
89.74 |
|
50 |
38.18 |
89.03 |
90.20 |
92.18 |
|
60 |
44.78 |
92.02 |
93.47 |
96.57 |
Note: SCD- surface solid dispersion of β- cyclodextrin
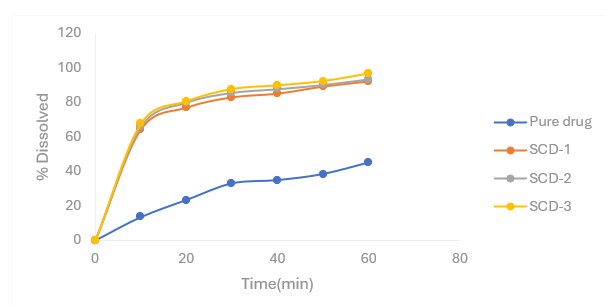
Figure 4.7 Percentage release profiles of Nifedipine SDs with β- cyclodextrin as carrier in 6.8 pH phosphate buffer
Spectral analysis by FTIR:
The spectrum analysis of the prepared solid dispersion of Nifedipine of SCD-3 is studied by FTIR. The resultant spectra were compared for any spectral changes. They were observed for the presence of characteristic peaks for the respective functional group in the compound. The pure drug and β-cyclodextrin were analysed by FTIR in same manner and were observed for the presence of respective peaks. The IR spectra of physical mixture showed in fig 4.8 matched with those of drug and β-cyclodextrin when superimposed. The spectrum of the surface solid dispersion SCD-3 showed characteristic peaks of both drug and β-cyclodextrin.
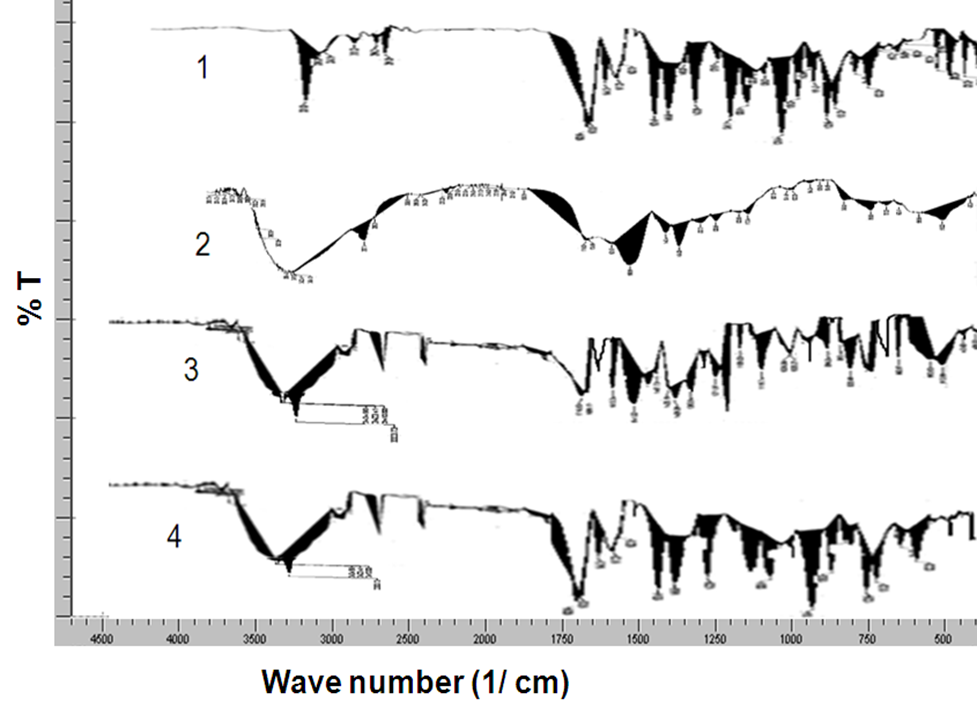
Figure 4.8 FTIR of Nifedipine
Surface morphology by SEM
The surface morphology of Nifedipine SSD on β-cyclodextrin (1:5) and PM were observed by using an analytical scanning electron microscope. The SEM of physical mixture as shown in fig.4.18 (A and B) showed dusting of drug powder on the carrier, while the SEM of surface solid dispersion as shown in fig.4.18 (C and D) showed a more porous nature of carrier with fine crystals of Nifedipine deposited on it when compared to physical mixture. This change in structure may be one of the causes for increase in dissolution rate.

Fig no 4.18: SEM photographs of A- PCD-3 at 750X, B- PCD-3 at 1000X, C- SCD-3 at 750X and D- SCD-3at 1000X.
Preparation And Evaluation of Oral Dispersible Tablets of Nifedipine:
Oral dispersible tablets were prepared using optimized surface solid dispersion of Nifedipine SCD-3 with different disintegrants. The SSDs were thoroughly mixed with microcrystalline cellulose, mannitol, silicified microcrystalline cellulose, sodium saccharin and the different disinetgrants like kyron T-314 (NFK), crospovidone (NFCP). The tablets were also prepared without disintegrants (NFS) and compared with the above tablets. The results of evaluation tests performed on the formualtions prepared are reported in Table no 4.11. The results indicated that batch of tablets complied with the official specifications. The dissolution profile of the tablets prepared using Nifedipine SCD-3 is compared with pure drug (table no 4.12 and fig no 4.19).
Table no 4.11: Evaluation of different formulations of oral dispersible tablets of Nifedipine:
|
Formulations |
Weight variation (mg + SD) |
Hardness (kg/cm2+ SD) |
Friability (% + SD) |
Disintegration time (seconds) |
|
NFS |
200.42 + 0.34 |
3.46 + 0.10 |
200.07+ 0.34 |
185 |
|
NFK |
200.32 + 0.40 |
3.32 + 0.12 |
200.1 + 0.38 |
25 |
|
NFCP |
200.18 + 0.27 |
3.35 + 0.12 |
200.06 + 0.30 |
30 |
Table no 4.12: Percentage release profiles of oral dispersible tablets (NFS, NFK, NFCP) of Nifedipine prepared with SCD-3:
|
Time (minutes) |
Pure drug |
NFS |
NFK |
NFCP |
|
0 |
0 |
0 |
0 |
0 |
|
10 |
13.44 |
65 |
74.83 |
71.22 |
|
20 |
23.24 |
84 |
86.08 |
89.39 |
|
30 |
32.81 |
90.83 |
92.94 |
92.39 |
|
40 |
34.66 |
93.05 |
95.40 |
95.12 |
|
50 |
38.18 |
95.12 |
97.03 |
97.58 |
|
60 |
44.78 |
98.02 |
99.78 |
99.50 |
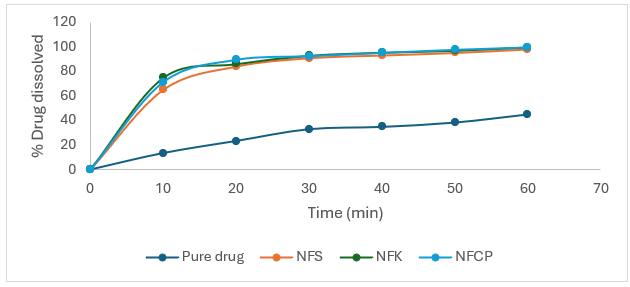
Fig no 4.19: Percentage release profiles of oral dispersible tablets (NFS, NFK, and NFCP) of Nifedipine prepared with SCD-3.
The above results indicated that SSDs of Nifedipine prepared to enhance dissolution rate could be successfully incorporated into a tablet without any much difference in the release properties between the prepared tablets and those of the SSDs. Tablets maintained the release behavior in showing the enhancement of drug release when compared to that of pure drug and tablets met all the required official specifications. The tablets with kyron T-314 (NFK) as disintegrant showed good disintegration and immediate availability of Nifedipine when compared to tablets prepared with crospovidone (NFCP) the tablets without any disintegrant (NFS) showed more disintegration time and slow release in the first 10 minutes indicating requirement of external disintegrants.
CONCLUSION
Rationale of the present study is to improve the dissolution rate of poorly soluble drug utilizing the approach of solid dispersion prepared by solid dispersion (SD) technique with different hydrophilic, insoluble, porous carriers like croscarmellose sodium, sodium starch glycolate and cyclodextrin. The drug selected for this study is Nifedipine. It was envisaged that SD technique may prove successful, since these drugs are poorly water soluble but show good bioavailability (BCS classification of class-II). The use of carriers is necessary to decrease the particle size of the drug to molecular level and thus enhancing the dissolution by increasing the surface area of the drug. Preliminary studies were done by preparing physical mixtures with different ratios of drug and carrier using simple blending method. They were characterized by the dissolution study to observe the ratio showing the enhanced dissolution. Nifedipine with β-cyclodextrin at 1:5 ratio of solid dispersion showed enhanced dissolution than other carriers when compared to pure drug which may be due to drug getting coated on the carrier. Then solid dispersions of Nifedipine were prepared with different carriers at different ratios by solvent evaporation method. The prepared SSDs were characterized by in vitro dissolution to analyze the improvement in the drug release compared to that of the pure drug and physical mixture. It was observed that solid dispersions of Nifedipine showed enhanced dissolution of Nifedipine due to the drug getting coated on the surface of carrier in molecular level and thereby enhanced surface area of the drug. It was also observed that as the carrier ratio was increased, the dissolution got increased. The drug: carrier ratio of 1:5 SDs showed enhanced dissolution than 1:1 ratio SDs due to the more carrier getting available for coating the drug. The SDs of Nifedipine with β-cyclodextrin showed greater enhancement of dissolution than other carriers when compared to pure drug and physical mixtures. Nifedipine with β-cyclodextrin showed enhanced dissolution followed by croscarmellose sodium, SSG. Based on the dissolution profiles of SDs, the SD which showed enhanced dissolution was optimized. Nifedipine with β-cyclodextrin at 1: 5 ratio SD showed enhanced dissolution and thereby optimized. The optimized solid dispersion was compressed into oral dispersible tablets successfully by using different disintegrants like kyron T-314 and crospovidone croscarmellose sodium. These tablets were evaluated for assay, content uniformity, hardness, friability, disintegration and in vitro dissolution. The results indicated that the tablets complied with the official specifications. The dissolution profiles of the tablets were compared with the pure drug. The tablets showed enhanced dissolution compared to that of the pure drug. The tablets prepared with kyron T-314 as disintegrant gave good disintegration and immediate availability of the drug than compared to other disintegrants. In conclusion, it can be stated that the objective of the study has been achieved. Surface solid dispersion technique was successful in improving the dissolution rate of poorly soluble drug Nifedipine. Hydrophilic, insoluble, porous carrier kyron T-314 was successful in improving the dissolution rate of Nifedipine by SSD technique. Tablets with improved dissolution rate can be prepared by using SSD.
REFERENCES


M. Sudhakar, G. Jyothi*, A. Akshaya, A. Navanitha, A. Hanuma, Amtul Mateen, Enhancement of Solubility of Nifedipine by Solid Dispersion Technique, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 1313-1340. https://doi.org/10.5281/zenodo.15844938
 10.5281/zenodo.15844938
10.5281/zenodo.15844938