
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1 KTN College of Pharmacy, Chalavara, Palakkad, Kerala
2 College of Pharmaceutical Sciences, Govt. Medical College, Kozhikode, Kerala.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder of the central nervous system. It is the most common cause of dementia in the elderly population in which there is a progressive deterioration of intellectual and social functions. No cure has yet been discovered for Alzheimer’s disease. But symptomatic therapies are widely available and they offer significant relief to patients and benefits to caregivers in terms of reduced care burden. Rivastigmine is a slow reversible inhibitor of acetylcholinesterase. It improves cognition, activities for daily living and global function. It is presently available orally in the form of tablets and capsules. Older people and bedridden patients find it difficult to swallow conventional tablets or capsules. In these patients, medication compliance and therapeutic effect could be improved by taking fast dissolving tablets. Fast dissolving tablets dissolve or disintegrate in the oral cavity without the need of water. It must include substances to mask the bitter taste of the active ingredient. This when swallowed by the patient, the drug get absorbed from the oral cavity, pharynx and esophagus as the saliva passes down into the stomach. So, it speeds up the dissolution, absorption and onset of action. Thus the bioavailability of drug is more when compared to those observed from conventional tablets dosage form. The fast dissolving solid dosage form turns into a soft paste or liquid form for easy swallowing, and thus it is free of risk of choking.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder of the central nervous system. It is the most common cause of dementia in the elderly population in which there is a progressive deterioration of intellectual and social functions, memory loss, personality changes and inability for self care. No cure has yet been discovered for AD. But symptomatic therapies are widely available and they offer significant relief to patients and benefits to caregivers in terms of reduced care burden.1 The agents recommended for the symptomatic treatment of mild to moderate AD include anticholinesterases such as rivastigmine, donepezil and galantamine and NMDA receptor antagonist such as memantine. Rivastigmine is a slow reversible inhibitor of acetyl cholinesterase (AChE) and butyrylcholinesterase (BuChE), while donepezil and galantamine are considered as AChE-selective, rapidly-reversible inhibitors. Cognitive improvement monitored by the Alzheimer’s Disease Assessment scale-cognitive subscale (ADAS- Cog) is greatest for rivastigmine relative to other anticholinesterase inhibitors. Treatment of mild to moderate AD using rivastigmine improves cognition, activities of daily living and global function.2 Rivastigmine is available as its soluble form rivastigmine tartrate. It is presently available orally in the form of tablets and capsules3. Older people and bedridden patients find it difficult to swallow conventional tablets or capsules. In these patients, medication compliance and therapeutic effect could be improved by taking fast dissolving tablets. United States Food and Drug Administration (FDA) defined fast dissolving tablet (FDT) as “a solid dosage form containing medicinal substance or active ingredient which disintegrate rapidly usually within a matter of seconds when placed upon the tongue.” According to European Pharmacopoeia, “Fast dissolving tablets should disperse/ disintegrate in the oral cavity within three minutes.” Fast dissolving tablets are also known as mouth-dissolving tablets, melt-in mouth tablets, orodispersible tablets, rapimelts, porous tablets, quick dissolving tablets, etc. Fast dissolving tablets dissolve or disintegrate in the oral cavity without the need of water.3,4 Fast dissolving tablets must include substances to mask the bitter taste of the active ingredient. This masked active ingredient is then swallowed by the patient's saliva along with the soluble and insoluble excipients. Some drugs get absorbed from the oral cavity, pharynx and esophagus as the saliva passes down into the stomach. The time for disintegration of fast disintegrating tablets is generally considered to be less than one minute. So, faster the dissolution, faster the absorption and onset of action. Thus, the bioavailability of drug is more in FDT when compared to those observed from conventional solid dosage form. Moreover, the fast dissolving solid dosage form turns into a soft paste or liquid form for easy swallowing and thus it is free of risk of choking.5 So, in the present study an attempt was made to formulate a fast dissolving tablet of rivastigmine tartrate by using various superdisintegrants and thereby increase its bioavailability and improve patient compliance.
MATERIALS AND METHODS
MATERIALS:
Rivastigmine tartrate, Microcrystalline cellulose, Sodium starch glycollate, Crospovidone, Croscarmellose sodium, Sucralose, Mannitol, Talc, Magnesium stearate, Potassium dihydrogen orthophosphate, Sodium hydroxide pellets.
METHODS:
a. Analysis of drug6
Physical properties:
Physical appearance of drug was determined by using various organoleptic properties and checked whether it complies with the specifications as prescribed in British Pharmacopoeia(2015). The melting point of rivastigmine tartrate was determined by capillary method. A small amount of the drug was placed in a capillary tube and the temperature at which the sample melted was noted with the help of a melting point apparatus.
Infrared spectral analysis:
The FT-IR spectra of rivastigmine tartrate was taken from Shimadzu FT-IR spectrophotometer. The FT-IR spectrum was prepared using KBr discs (spectroscopic grade). The drug was finely ground with KBr to prepare pellets under a hydraulic pressure of 600 psi and a spectrum was scanned in the wavelength range of 400-4000cm-1. The spectrum was then compared with the reference spectrum of rivastigmine tartrate.
Solubility:
Solubility of rivastigmine tartrate was determined in different solvents. An excess quantity of the drug was mixed with 10ml of each solvent in test-tubes and shaken for 24 hours at room temperature.
Saturation solubility: Saturation solubility of the drug was determined by dissolving the excess amount of the drug in 10ml of pH 6.8 phosphate buffer. The solution was filtered by using Whatmann filter paper and diluted if needed. The amount of drug dissolved was analysed by UV spectrophotometer.
b. Analysis of excipients7
Sodium starch glycollate
Identification test: To 5 ml of a 2% w/v dispersion in water 0.05 ml of 0.005M iodine was added.
Croscarmellose sodium
Identification test: 1g of croscarmellose sodium was mixed with 50 ml of water. 1 ml of the mixture was transferred to a test tube, added 1ml of water and 0.05ml of a freshly prepared 4%w/v solution of alpha naphthol in methanol. Inclined the test tube and added carefully 2ml of sulphuric acid down the sides.
Crospovidone
Identification test: Suspended 1g in 10ml of water , added 0.1 ml of 0.1M iodine and shaken for 30 seconds. Then added 1ml of starch solution and shaken.
Mannitol
Identification test: To 1 ml of a saturated solution 0.5 ml of ferric chloride test solution was added followed by addition of 0.25 ml of sodium hydroxide solution and shaken well.
Microcrystalline cellulose
Identification test: To 50 mg of microcrystalline cellulose, 2 ml of iodine solution was added and allowed to stand for 5 minutes, removed the excess reagent with the aid of a filter paper and added 1 or 2 drops of sulphuric acid (66% v/v).
c. Drug excipient compatibility study
Drug excipient compatibility was determined by Shimadzu FT-IR spectrophotometer. Drug and the excipients used were mixed uniformly and the spectrum was taken. The FT- IR spectrum was prepared using KBr discs (spectroscopic grade). The drug excipient mixture was finely ground with KBr to prepare pellets under a hydraulic pressure of 600 psi and a spectrum was scanned in the wavelength range of 400-4000cm-1. The spectrum was then compared with the spectrum of rivastigmine tartrate.
Preparation of calibration curve of rivastigmine tartrate
Calibration curve of rivastigmine tartrate was prepared in pH 6.8 phosphate buffer.
Preparation of pH 6.8 phosphate buffer7 :
Preparation of 0.2M potassium dihydrogen phosphate:
Dissolved 27.218g of potassium dihydrogen phosphate in 1000 ml water.
Preparation of 0.2M NaOH:
Dissolved about 8g of sodium hydroxide in 1000ml water.
Preparation of pH 6.8 phosphate buffer:
Placed 50ml of 0.2M potassium dihydrogen phosphate in 200 ml volumetric flask. Added 22.4 ml of 0.2M of sodium hydroxide and made up the volume with water.
Determination of λmax of rivastigmine tartrate:
The λmax of rivastigmine tartrate was determined in the UV range 200-400nm by using UV spectrophotometer.
Preparation of calibration curve of rivastigmine tartrate
100mg of rivastigmine tartrate was weighed accurately and dissolved in pH 6.8 phosphate buffer in a100ml volumetric flask and was made to 100ml with the phosphate buffer. A stock solution of 1000µg/ml was obtained. From this 1, 2, 3, 4, 5 ml solutions was pipetted out to a 10ml of volumetric flask and made up the volume with pH 6.8 phosphate buffer to produce solutions in the range 100-500 µg/ml.
Fast dissolving tablets of rivastigmine tartrate was prepared by direct compression method. Different concentration of superdisintegrants sodium starch glycollate, crospovidone and croscarmellose (3%, 4%, 5%, 6%) was used to prepare different batches of fast dissolving tablets (F1-F12). Compositions of various formulations are shown in table. All the ingredients were weighed as given in the table. All the ingredients without magnesium stearate and talc were mixed uniformly followed by passing through sieve no. 100. Then magnesium stearate and talc were added and mixed uniformly. The prepared powder blend was evaluated for precompression parameters like bulk density, tapped density, angle of repose, compressibility index and Hausner’s ratio. After evaluation of powder blend the mixture were compressed using Cadmach tablet punching machine. The total weight of the formulation was maintained 150mg.
Table 1 Composition of different batches of fast dissolving tablets of rivastigmine tartrate
|
Ingredients (mg/ tablet) |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
F11 |
F12 |
|
Rivastigmine tartrate |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
|
Mannitol |
103.5 |
102 |
100.5 |
99 |
103.5 |
102 |
100.5 |
99 |
103.5 |
102 |
100.5 |
99 |
|
Microcrystalline cellulose |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
|
Sodium starch Glycollate |
4.5 |
6 |
7.5 |
9 |
- |
- |
- |
- |
- |
- |
- |
- |
|
Crospovidone |
- |
- |
- |
- |
4.5 |
6 |
7.5 |
9 |
- |
- |
- |
- |
|
Croscarmellose |
- |
- |
- |
- |
- |
- |
- |
- |
4.5 |
6 |
7.5 |
9 |
|
Sucralose |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
3 |
|
Talc |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
|
Magnesium stearate |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
|
Total |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
3. Evaluation of fast dissolving tablets
a. Precompression studies10
Bulk density
It is the ratio of total mass of powder to the bulk volume of powder. It was measured by pouring about 10gm of powder into a 50ml measuring cylinder and initial volume was noted. This initial volume was called the bulk volume. From this the bulk density was calculated according to the formula mentioned below. It is expressed in gm/ml.
ρb = M/Vb
Where, M is mass of the powder
Vb is bulk volume of powder
Tapped density
The measuring cylinder containing 10gm of powder was tapped until a constant weight was obtained . The minimum volume (Vt) occupied in the cylinder was measured. The tapped density was calculated using the following formula,
ρt=M/Vt
where, M is mass of powder
Vt is tapped volume of the powder
Carr’s compressibility index
The simplest way of measurement of free flow of powder is compressibility. The compressibility index of the granules was determined by Carr’s compressibility index which is calculated by using the following formula,
C= ( ρt - ρb / ρt ) ×100
Where, ρt is the tapped density
ρb is the bulk density
Hausner’s ratio
Hausner’s ratio is an indirect index of ease of powder flow. It is calculated by the following formula
Hausner’s ratio = ρt /ρb
Where, ρt is tapped density
ρb is bulk density.
Table 2 Scale of flowability
|
Flow character |
Carr’s index |
Hausner’s ratio |
|
Excellent |
≤ 10 |
1.00-1.11 |
|
Good |
11-15 |
1.12-1.18 |
|
Fair |
16-20 |
1.19-1.25 |
|
Passable |
21-25 |
1.26-1.34 |
|
Poor |
26-31 |
1.35-1.45 |
|
Very poor |
32-37 |
1.46-1.59 |
Angle of repose
Angle of repose (α) was determined using fixed funnel method. The blend was poured through a funnel that is kept at a height (h) of 2cm above the horizontal surface. The radius of the heap (r) was measured and angle of repose was calculated.
α= tan-1 (h/r)
where, h is the height of the heap
r is the radius of the heap
Table 3 Flow property and corresponding angle of repose
|
Angle of repose |
Type of flow |
|
<25 |
Excellent |
|
25-30 |
Good |
|
30-40 |
Passable |
|
>40 |
Very poor |
b. Postcompression studies11
Tablet thickness
Thickness of tablets were determined by using screw guage . Five tablets from each batch were taken and an average value was calculated. It was measured in mm.
Weight variation
Ten tablets were taken and their weight was determined individually and collectively on a weighing balance. The average weight was determined from the collective weight.
Hardness
Hardness indicates the ability of a tablet to withstand mechanical stress while handling. The hardness of tablet was measured by Pfizer hardness tester. It was measured in terms of Kg/cm².
Friability
Friability of the tablets was determined using Roche friabilitor. This device subjects the tablets to the combined effect of abrasions and shock in a plastic chamber revolving at 25 rpm and dropping the tablets at a height of 6 inches in each revolution. Preweighed sample of tablets (10) was placed in the friabilator and were subjected to 100 revolutions. Tablets were dusted using a soft muslin cloth and reweighed. The friability (f) is given by the formula.
f = (1- W0 / W) × 100
Where, W0 is weight of the tablets before the test
W is the weight of the tablet after the test.
Wetting time
Five circular tissue papers of 10 cm diameter was placed in a petridish with a 10 cm diameter Ten ml of water-containing a water-soluble dye (amaranth), was added to petridish. A preweighed tablet was carefully placed on the surface of the tissue paper. The time required for water to reach upper surface of the tablet was noted as a wetting time. It was measured in seconds.
Water absorption ratio
The wetted tablet was then weighed. Water absorption ratio R, was determined using the following equation:
R = 100 X Wa-Wb/Wb
Where, Wb is the weight of tablet before water absorption
Wa is the weight of tablet after water absorption.
Content uniformity
5 tablets were selected, powdered in a mortar and the average weight was taken. It was diluted in a suitable amount of phosphate buffer and was assayed by UV spectrophotometer at 263.1 nm.
Modified disintegration test23
As described in pharmacopoeia, tablets are placed in the disintegration tubes and time is noted. FDTs should disintegrate within 1 minute without leaving any residue on the screen. The conventional test employs a volume of 900 ml of distilled water compared to the volume of saliva in humans, which is limited to a few ml. The disintegration rate obtained from conventional test does not appear to reflect the actual disintegration rate in human mouth. The amount of saliva in the mouth is limited and no tablet disintegration test was found in USP and IP to simulate in vivo conditions. . To overcome these problems, several new methods have been proposed. A modified version of the simple but novel method was used here to determine disintegration time of the tablets. A cylindrical vessel was used in which 10-mesh screen was placed in such way that only 4 ml of disintegrating medium would be placed below the sieve. To determine disintegration time, 6ml of pH 6.8 phosphate buffer, was placed inside the vessel in such way that 4ml of the media was below the sieve and 2ml above the sieve. Tablet was placed on the sieve and the time at which all the particles pass through the sieve was taken as disintegration time of the tablet.
Invitro dissolution studies39
In vitro dissolution studies of the formulated tablets were studied in 50ml pH 6.8 phosphate buffer. A small dissolution volume was taken as the drug’s linearty range was found to be within 100-500µg/ml and also due to its small dose and high solubility in the dissolution medium. The temperature was maintained at 370C ± 0.5. The rotation speed was 50 rpm. 2.5 milliliters of aliquot were withdrawn at predetermined time intervals. The medium was replaced with 2.5 mL of buffer each time. Sample was analyzed by using UV spectrophotometer at 263.1nm.
c. Evaluation of optimized formulation33
Stability study
The optimized tablet was analysed for physical appearance, drug content and drug release at 0- and 30-days interval. Tablets were kept for 30 days at room temperature and was analyzed by UV spectrophotometer at 263.1 nm.
RESULTS AND DISCUSSIONS
1. Preformulation studies
a. Analysis of drug
Physical properties
The physical properties of the drug complied with the specifications given in British Pharmacopoeia (2015). Therefore, it was confirmed that the drug was rivastigmine tartrate.
Infrared spectral analysis
The IR spectra of rivastigmine tartrate showed no differences in the major peak when compared to the reference spectra of rivastigmine tartrate. It showed C-H (str) at 2934 cm- 1, C-N (str) at 2876 cm-1, C=O (str) at 1654 cm-1 and C=C (str) at 3045 cm-1
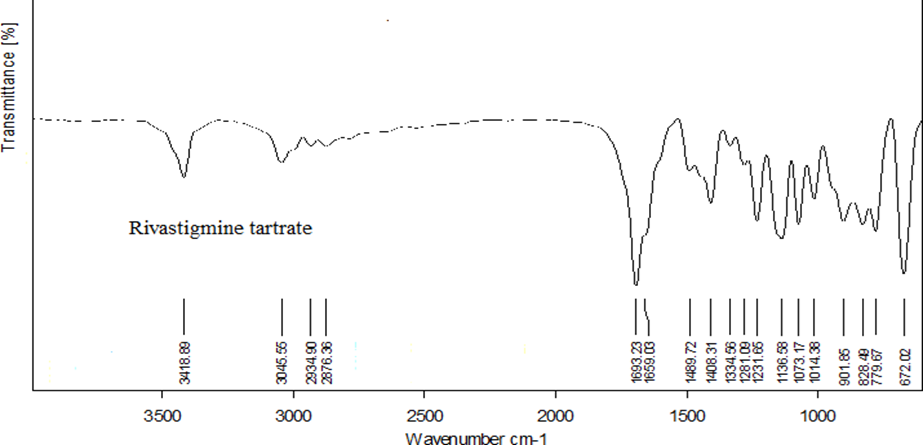
Fig.1 Reference spectra of rivastigmine tartrate
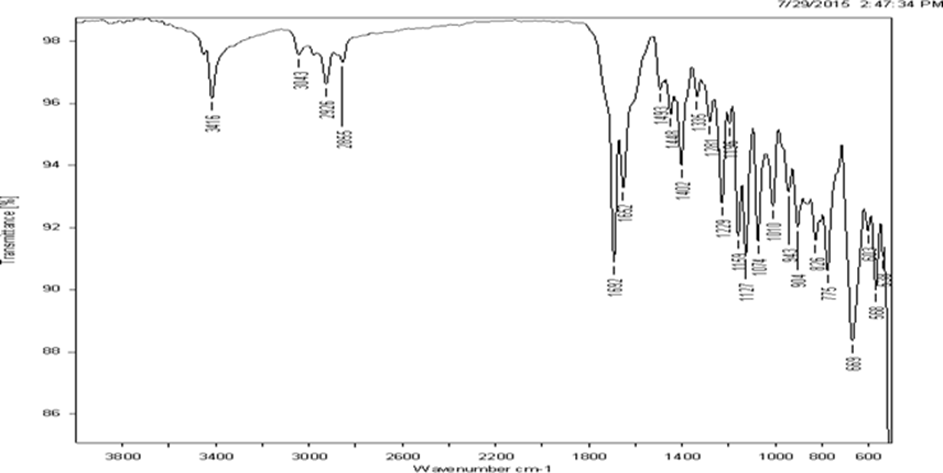
Fig.2 FT-IR spectrum of rivastigmine tartrate
Solubility
The solubility of rivastigmine tartrate was studied in different solvents and the results were in compliance with the standards specified in BP (2015).
Table 4 Solubility analysis of rivastigmine tartrate in different solvents
|
Solvents |
Solubility |
|
Water |
Soluble |
|
Methanol |
Soluble |
|
Ethyl acetate |
Slightly soluble |
Saturation solubility
The saturation solubility of rivastigmine tartrate in pH 6.8 phosphate buffer was found to be 350mg/ml.
b. Analysis of excipients
The various identification tests specified as per I.P (2014) for each excipients were carried out and thereby its identity was confirmed.
Sodium starch glycolate
Identification test: When iodine was added to a 5 ml of a 2% w/v dispersion in water a dark blue colour was produced.
Croscarmellose sodium
Identification test: 1g of croscarmellose was mixed with 50 ml of water. Then transferred 1ml of the mixture to a test tube and added 1ml of water and 0.05ml of a freshly prepared 4%w/v solution of alpha naphthol in methanol. The test tube was inclined and a 2ml of sulphuric acid was poured down the side. A lower layer was formed which confirmed the presence of croscarmellose sodium.
Crospovidone
Identification test: Iodine was added to a 1g of crospovidone suspended in 10ml of water. When starch solution was added to the mixture no blue colour developed which confirmed the presence of crosspovidone.
Mannitol
Identification test: When 0.5 ml of ferric chloride test solution followed by 0.25 ml of sodium hydroxide solution was added to a saturated solution of mannitol a clear solution was obtained which remained clear on further addition of sodium hydroxide solution.
Microcrystalline cellulose
Identification test: When 2 ml of iodine solution was added to microcrystalline cellulose followed by addition of 1 or 2 drops of sulphuric acid (66% v/v); a blue-purple colour was produced.
c. Drug excipient compatibility studies
IR spectrum of the pure drug was compared with the IR spectrum of drug excipient mixture. All the characteristic peaks that appeared in the IR spectra of drug were present in the spectra of drug excipient mixture indicating that there was no drug excipient interaction.
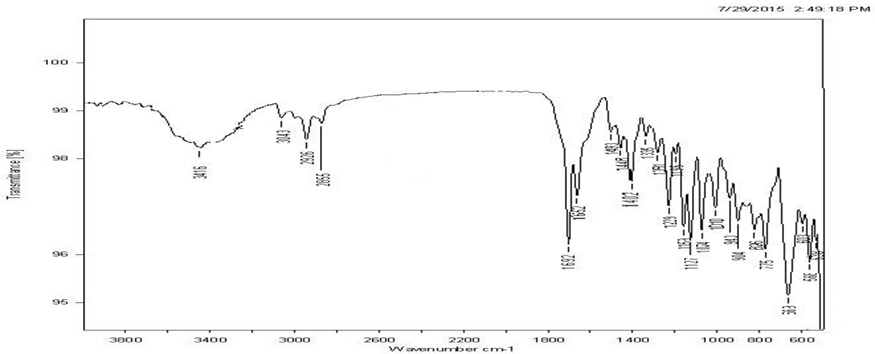
Fig. 3 FT-IR Spectra of mixture of drug, sodium starch glycollate and other excipients
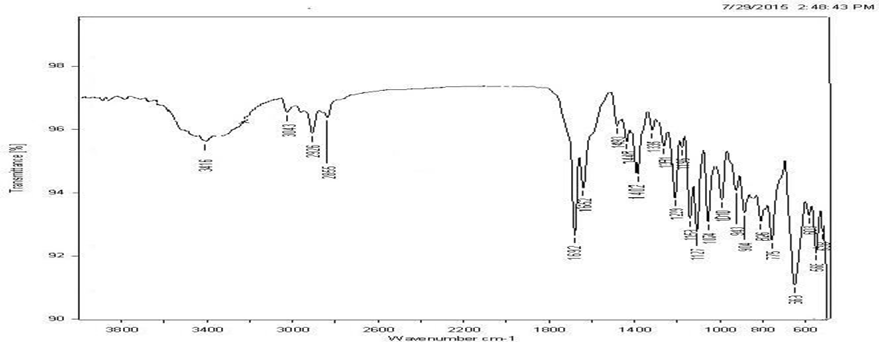
Fig. 4 FT-IR Spectra of mixture of drug, crospovidone and other excipients
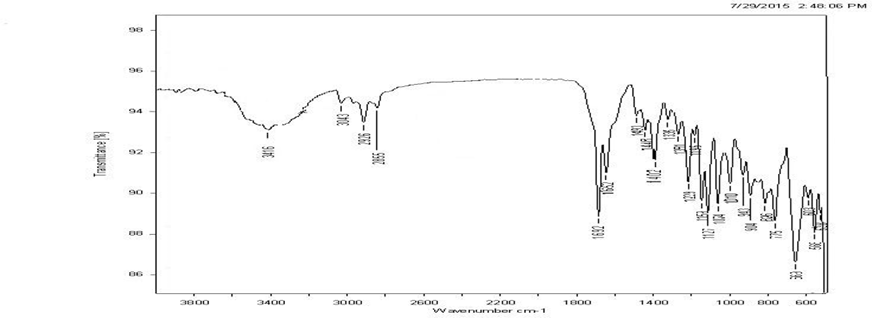
Fig. 5 FT-IR Spectra of mixture of drug, croscarmellose and other excipients
Preparation of calibration curve of rivastigmine tartrate in pH 6.8 phosphate buffer
The λmax of rivastigmine tartrate in pH 6.8 phosphate buffer was found to be 263.1nm. The calibration curve of rivastigmine tartrate in phosphate buffer was plotted with concentration of drug solution in µg/ml on x- axis and absorbance on y-axis. The linearity was found in the concentration range of 100-500 µg/ml with a regression coefficient value of 0.997.
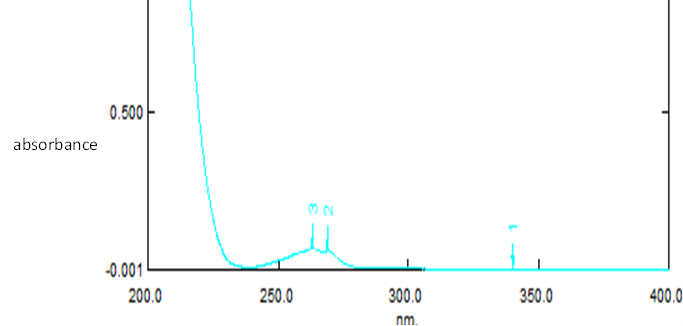
Fig. 6 UV spectra of rivastigmine tartrate in pH 6.8 phosphate buffer
Table 5. Calibration plot of rivastigmine tartrate in pH 6.8 phosphate buffer
|
Concentration (µg/ml) |
Absorbance |
|
0 |
0 |
|
100 |
0.1700 |
|
200 |
0.3229 |
|
300 |
0.4559 |
|
400 |
0.5920 |
|
500 |
0.7613 |
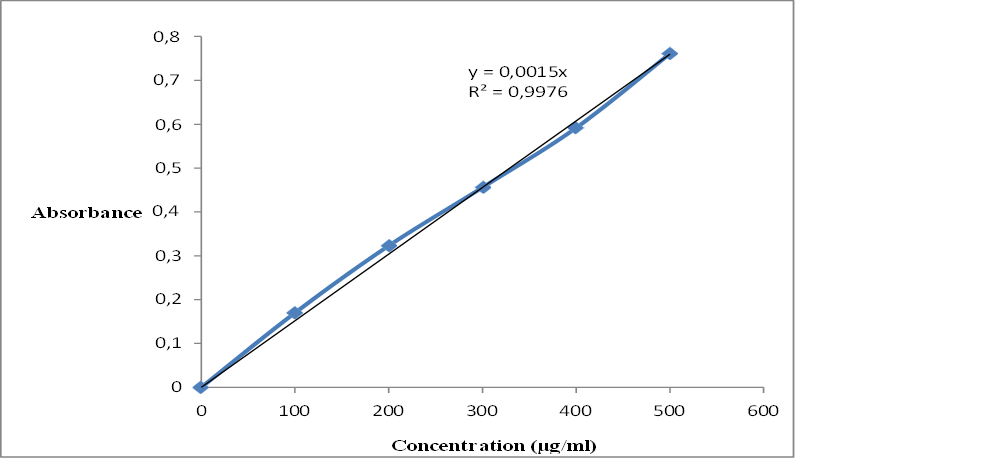
Fig 7. Calibration curve of rivastigmine tartrate in pH 6.8 phosphate buffer
2. Evaluation of the fast dissolving tablets
a. Precompression studies
Bulk density
All the formulations were found to comply with the limits specified in the official pharmacopoeia. The bulk density of the powder blend varied from 0.4113 to 0.4696 g/cm3.
Tapped density
All the formulations were found to comply with the limits specified in the official pharmacopoeia. The tapped density was found in the range from 0.4480 to 0.5547 g/cm3 .
Carr’s compressibility index
Carr’s compressibility index for the formulations was within the range of 8.1510 to 15.2633 % which indicated that the powder exhibited good compressibility.
Hausner’s ratio
Hausner’s ratio for the formulations varied from 1.0889 to 1.1791. Hausner’s ratio in the range1.00 -1.11 indicated excellent flow property and in the range 1.12-1.18 showed good flow property. As all the formulations were within this range it indicated that it has good to excellent flow property.
Angle of repose
Angle of repose for all the formulations ranged from 27?.243’ to 30?.58’. Angle of repose in the range 25-30 indicates good flow property. As all the formulation where within this range, the powder blend exhibited good flow property.
Table 6. Precompression studies of F1, F2, F3, F4 (Mean±SD)
|
Evaluation |
F1 |
F2 |
F3 |
F4 |
|
Bulk density (g/cm3) |
0.4435 ± 0.039 |
0.4548 ± 0.021 |
30.4295 ± 0.027 |
0.4113 ± 0.019 |
|
Tapped density (g/cm3) |
0.4866 ±0.047 |
0.5006 ± 0.025 |
0.4707 ±0.0334 |
0.4480 ±0.0242 |
|
Carr’s index (%) |
8.8016 ±0.890 |
9.4166 ± 0.344 |
8.573 ± 0.684 |
8.1510 ± 0.580 |
|
Hausner’s ratio |
1.0966 ± 0.010 |
1.1005 ±0.004 |
1.0939 ±0.007 |
1.0889 ±0.006 |
|
Angle of repose (?) |
27.653 ± 0.472 |
27.243 ± 0.463 |
26.850 ± 0.290 |
26.563 ±0.285 |
TABLE 7. Precompression studies of F5, F6, F7, F8 (Mean±SD)
|
Evaluation |
F5 |
F6 |
F7 |
F8 |
|
Bulk density (g/cm3) |
0.4488±0.0303 |
0.4306±0.0400 |
0.4586±0.0522 |
0.4515±0.0500 |
|
Tapped density (g/cm3) |
0.4935±0.0361 |
0.4783±0.0412 |
0.5118±0.0517 |
0.5032±0.0583 |
|
Carr’s index (%) |
9.0251±0.5215 |
9.9886±1.8096 |
10.5786±1.2997 |
10.3936±1.4692 |
|
Hausner’s ratio |
1.0990±0.0065 |
1.1110±0.0227 |
1.1170±0.0167 |
1.1154±0.0186 |
|
Angle of repose (?) |
28.07 ± 0.3202 |
28.39 ± 0.3251 |
28.72 ± 0.3300 |
29.15 ± 0.4986 |
Table 8 Precompression studies of F9, F10, F11, F12 (Mean±SD)
|
Evaluation |
F9 |
F10 |
F11 |
F12 |
|
Bulk density (g/cm3) |
0.4446±0.0508 |
0.4346±0.0519 |
0.4628±0.0338 |
0.4696±0.0262 |
|
Tapped density (g/cm3) |
0.5031±0.0503 |
0.5142±0.0679 |
0.5380±0.0453 |
0.5547±0.0313 |
|
Carr’s index (%) |
11.7603±1.5677 |
13.3636±1.4613 |
13.8966±0.8905 |
15.2633±1.1884 |
|
Hausner’s ratio |
1.1320±0.0169 |
1.1520±0.0169 |
1.1591±0.0101 |
1.1791±0.0257 |
|
Angle of repose(?) |
29.73 ±0.7057 |
29.41 ± 0.9240 |
30.32 ± 0.5632 |
|
Postcompression studies
Thickness
Thickness of the formulated tablets was measured using screw gauge and the values ranged from 2.4- 2.7 mm.
Weight variation
The weight variation was evaluated and found to be within the range of 146.9 -149.1mg. All the formulations passed the weight variation test as it was within the range (7.5%) specified in I.P (2014).
Hardness
Hardness of the tablets of all the formulations varied from 3.1 to 3.8 kg/cm2 which indicates that the tablets have sufficient hardness for easy handling. But it should not be too hard to prevent easy disintegration of the tablet.
Friability
Friability of the tablets was found to be within the range of 0.3923 to 0.7980 % and is well within the approved range of < 1% as per IP. It indicates the tablets ability to withstand the mechanical stress during packing and shipping.
Drug content
Drug content of all the formulations was found to be within the limit specified in I.P. (85- 115%) and are given in the table. It ranged from 96.5- 100.1%.
Wetting time
Wetting time is closely related to the inner structure of the tablet. Wetting time was found in the range of 10 to 15 seconds. It was found to be least for F3 containing 5% sodium starch glycolate and maximum for F12 containing 6% crosscarmellose. Smaller wetting time of sodium starch glycolate may be due to its rapid uptake of water. Wetting time of 6% croscarmellose may be more as it formed a gel that inhibits water penetration into the tablet core.
Water absorption ratio
Water absorption ratio of all the formulations ranged from 47.3 to 70.5%. Water absorption ratio increased with decrease in wetting time. It was more for sodium starch glycollate. This may be due to its rapid water uptake.
Modified disintegration test
As FDTs intended to disintegrate in the saliva, a modified disintegration test was performed. All the formulations disintegrated within 17-30 seconds. The invitro disintegration time was least for tablets containing 5% sodium starch glycolate as superdisintegrant . This may be due to the rapid water uptake and easy swelling of sodium starch glycolate when compared to other superdisintegrants used.
Table 9 Post compression evaluation of F1, F2, F3, F4 (Mean±SD)
|
Evaluation |
F1 |
F2 |
F3 |
F4 |
|
Thickness (mm) |
2.5 ± 0.3 |
2.6± 0.2 |
2.5± 0.5 |
2.5± 0.3 |
|
Weight variation (mg) |
147.9 ±0.366 |
147.8±0.280 |
146.9 ±0.183 |
148.6± 0.524 |
|
Hardness (kg/cm2) |
3.7 ± 0.120 |
3.8±0.238 |
3.8±0.532 |
3.7±0.432 |
|
Friability (%) |
0.4256± 0.083 |
0.3923± 0.120 |
0.410±0.532 |
0.4765±0.580 |
|
Drug content (%) |
96.66±0.512 |
98.6±0.632 |
98.3±0.234 |
98.9±0.235 |
|
Wetting time (seconds) |
15.5±0.5 |
10.5±0.5 |
10±1 |
12±1 |
|
Water absorption ratio (%) |
50.5±0.523 |
68.7±0.502 |
70.5±0.212 |
59.5±0.132 |
|
Disintegration time (seconds) |
28±1 |
20.5±0.5 |
17.5±0.5 |
25±1 |
Table 10 Post compression evaluation of F5, F6, F7, F8 (Mean±SD)
|
Evaluation |
F5 |
F6 |
F7 |
F8 |
|
Thickness (mm) |
2.6± 0.2 |
2.7±0.3 |
2.5±0.2 |
2.5±0.1 |
|
Weight variation (mg) |
148.5±0.125 |
147.5±0.120 |
148.8±0.258 |
148.9±0.256 |
|
Hardness (kg/cm2) |
3.5±0.112 |
3.5±0.234 |
3.4±0.254 |
3.1±0.543 |
|
Friability (%) |
0.45±0.087 |
0.490±0.075 |
0.53±0.035 |
0.78±0.234 |
|
Drug content (%) |
96.8±0.532 |
98.5±0.432 |
98.3±0.243 |
100.1±0.234 |
|
Wetting time (seconds) |
15±1 |
12.5±0.5 |
12±1 |
13.5±2 |
|
Water absorption ratio (%) |
47.8±0.512 |
53.2±0.224 |
60.3±0.212 |
51.8±0.123 |
|
Disintegration time (seconds) |
30±1 |
23±1 |
19.5±0.5 |
26±1 |
Table 11 Post compression evaluation of F9, F10, F11, F12 (Mean±SD)
|
Evaluation |
F9 |
F10 |
F11 |
F12 |
|
Thickness (mm) |
2.4±0.4 |
2.5±0.1 |
2.4±0.3 |
2.6±0.2 |
|
Weight variation (mg) |
147.9±0.926 |
148.5±0.123 |
148.9±0.563 |
149.1±0.432 |
|
Hardness (kg/cm2) |
3.5±0.256 |
3.4±0.534 |
3.3±0.123 |
3.1±0.102 |
|
Friability (%) |
0.488±0.145 |
0.609±0.135 |
0.632±0.432 |
0.798±0.342 |
|
Drug content (%) |
98.6±0.456 |
97.2±0.435 |
96.5±0.324 |
98.9±0.369 |
|
Wetting time (seconds) |
12±1 |
11±1 |
13.5±0.5 |
15.5±0.5 |
|
Water absorption ratio (%) |
52.5±0.276 |
60.65±0.248 |
50.53±0.356 |
47.3±0.319 |
|
Disintegration time (seconds) |
24±1 |
20±1 |
26.5±0.5 |
30±1 |
In vitro dissolution studies
All the formulations initially showed an increase in drug release with an increase in super disintegrant concentration up to a limit. On further increase in concentration, the drug release decreased. The formulations containing sodium starch glycollate F1 (3%), F2 ( 4%), F3 (5%) and F4 (6%) showed drug release of 90%, 98.1% 99.8%and 90.6% respectively in 75 seconds. There was an increase in drug release upto 5% of sodium starch glycollate. This may be attributed to the rapid water uptake and easy swelling of the tablets due to the presence of sodium starch glycollate. At higher levels (above 5%), formation of a viscous gel layer by sodium starch glycollate might have formed a thick barrier for further penetration of the disintegration medium and hindered the disintegration or leakage of tablet contents. The formulations containing crospovidone F5 (3%), F6 ( 4%), F7 (5%) and F8 (6%) showed drug release of 72.9%, 96.5%, 97.81% and 87.4% at 75 seconds respectively. Crospovidone acts as a superdisintegrant by capillary or wicking action. Its decrease in drug release above 5% may be due to its difficulty to get soaked in water. The formulations containing croscarmellose sodium F9 (3%), F10 ( 4%), 11 (5%) and F12 (6%) showed drug release of 91.6%, 98.2%, 83.56% and 81.2% at 75 seconds respectively. At higher concentrations, croscarmellose sodium will form a gel that inhibits water penetration into the tablet core, thereby decreasing the release. Among the 12 formulations prepared with superdisintegrants sodium starch glycollate, crospovidone and croscarmellose, F3 (5% sodium starch glycollate) was the best one with the least wetting and disintegration times and maximum release of 99.8% in 75 seconds.
Table 12 Percentage cumulative drug release of F1, F2, F3, F4 ( Mean(%)±SD)
|
Time (seconds) |
F1 |
F2 |
F3 |
F4 |
|
0 |
0 |
0 |
0 |
0 |
|
15 |
43.3±0.123 |
67.7±0.274 |
70.8±0.644 |
42.3±0.345 |
|
30 |
60.3±0.234 |
76.27±0.573 |
83.4±0.554 |
55.4±0.274 |
|
45 |
64.2±0.543 |
84.27±0.453 |
87.5±0.523 |
63.5±0.382 |
|
60 |
78.9±0.265 |
94.9±0.388 |
99.1±0.552 |
84.1±0.332 |
|
75 |
90±0.345 |
98.1±0.543 |
99.8±0.643 |
90.6±0.395 |
|
90 |
94±0.224 |
98.5±0.447 |
98.9±0.337 |
93.3±0.283 |
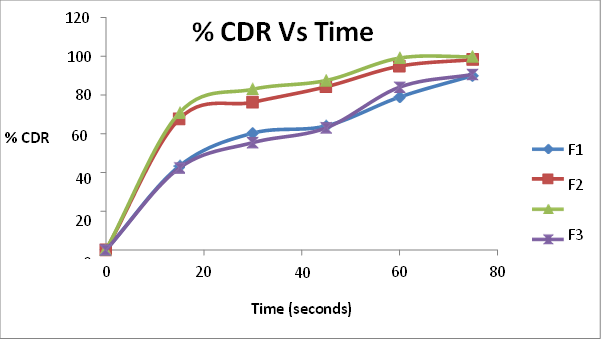
Fig. 8. Percentage cumulative drug release of F1, F2, F3, F4
Table 13 Percentage cumulative drug release of F5, F6, F7, F8 ( Mean(%)±SD)
|
Time (seconds) |
F5 |
F6 |
F7 |
F8 |
|
0 |
0 |
0 |
0 |
0 |
|
15 |
40.2±0.329 |
56.4±0.256 |
59.2±0.340 |
42.4±0.345 |
|
30 |
42.37±0.338 |
73.2±0.543 |
76.2±0.338 |
52.5±0l.254 |
|
45 |
51.7±0.281 |
85.2±0.432 |
86.84±0.289 |
72.0±0.287 |
|
60 |
67.7±0.284 |
93.2±0.537 |
94.25±0.394 |
83.62±0.124 |
|
75 |
72.9±0.765 |
96.5±0.228 |
97.81±0.129 |
87.4±0.256 |
|
90 |
85.2±0.445 |
98.1±0.112 |
98.9±0.543 |
94.9±0.654 |
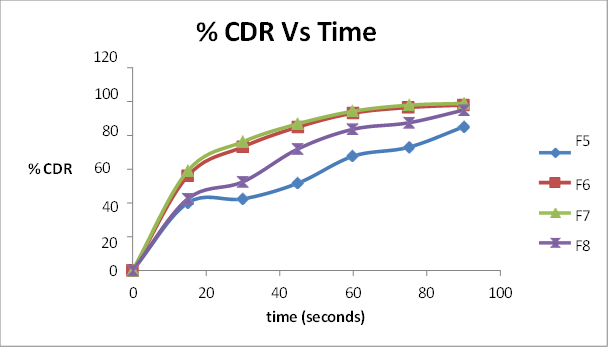
Fig.9 Percentage cumulative drug release of F5, F6, F7, F8
Table 14 Percentage cumulative drug release of F9, F10, F11, F12 ( Mean(%)±SD)
|
Time (seconds) |
F9 |
F10 |
F11 |
F12 |
|
0 |
0 |
0 |
0 |
0 |
|
15 |
42.37±0.235 |
43.1±0.467 |
41.57±0.381 |
39.5±0.385 |
|
30 |
50.12±0.543 |
64.6±0.345 |
51.57±0.128 |
47.69±0.537 |
|
45 |
64.58±0.654 |
71.12±0.224 |
58.08±0.673 |
55.47±0.882 |
|
60 |
75.66±0.638 |
94.82±0.543 |
72.03±0.453 |
70.3±0.443 |
|
75 |
91.66±0.629 |
98.2±0.187 |
83.56±0.233 |
81.2±0.213 |
|
90 |
93.5±0.189 |
98.6±0.285 |
91.52±0.123 |
88.9±0.118 |
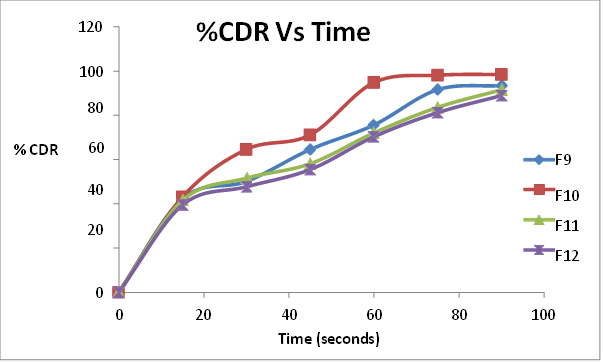
Fig.10 Percentage cumulative drug release of F9, F10, F11, F12
a. Evaluation of the optimized tablet
Stability study
Stability study carried out on the optimized tablet showed no significant change in the physical appearance, drug content, invitro release and other significant parameters of the tablet when compared to the formulation on the zeroth day.
Table 15. Stability study of the optimized formulation
|
|
0 days |
30 days |
|
Drug content |
98.3 |
97.5 |
|
% CDR at 75 sec |
99.8 |
98.54 |
CONCLUSION
Rivastigmine tartrate, an anticholineesterase drug is usually prescribed for the symptomatic treatment of AD. Due to the problem of dysphagia, patients suffering from AD find it difficult to swallow conventional solid dosage forms of rivastigmine tartrate. This may lead to ineffective therapy. As an attempt to overcome this problem, a fast dissolving tablet of rivastigmine tartrate was formulated to improve patient compliance and medication adherence. The FDTs are solid dosage forms which disintegrate or disperse in the saliva within few seconds. In this study, the FDTs of rivastigmine tartrate was prepared by using superdisintegrant approach by direct compression. Initially, the drug and other excipients were subjected to identification tests as specified in the official monograph so as to confirm its identity. The drug and excipients were uniformly blended and carried out the compatibility study and precompression evaluations. In order to ensure complete disintegration of the formulation in saliva, different superdisintegrants in varying concentrations (3%, 4%, 5% and 6%) were used. Postcompression studies which include thickness, weight variation, hardness, friability, and drug content were determined for all the formulations and found to be within the limits as prescribed in the official pharmacopoeia. Other tests carried out for FDTs include modified disintegration test, wetting time, water absorption ratio and invitro release studies. Wetting time and disintegration time decreased with increasing concentration of crospovidone, sodium starch glycollate and croscarmellose. A further increase in concentration of the superdisintegrants crospovidone and sodium starch glycollate beyond 5% resulted in a slight increase in wetting time and disintegration time. Similarly, the drug release was also found to be increasing, with increase in concentration of superdisintegrants. A 5% concentration of sodium starch glycollate showed a maximum release of 99.8% within 75 seconds. This may be due to the rapid water uptake and easy swelling of the tablets containing sodium starch glycollate. On the other hand, crospovidone showed a maximum release of 97.8% at 5 % concentration within 75 seconds. Its decrease in release may be due to its difficulty to get soaked in water. A 4% concentration of croscarmellose showed a maximum release of 98.2% in 75 seconds which may be comparable with the release of tablet containing 5% sodium starch glycollate. As the mechanical strength of 4% croscarmellose containing formulation was less when compared to the formulation containing 5 % sodium starch glycollate, the latter was preferred as the optimum formulation. It was subjected to further evaluation of short term stability studies and was found that the tablet exhibited sufficient stability.
REFERENCES


Labeeba Paduvan Padath, Anju P, Formulation, Evaluation and Optimization of Fast Dissolving Tablets of Rivastigmine Tartrate, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 1122-1140. https://doi.org/10.5281/zenodo.15838200
 10.5281/zenodo.15838200
10.5281/zenodo.15838200