
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,3,4,5,6UG Scholar, Rashtriya College of Pharmacy, Hatnoor, Kannad, Chh. Sambhajinagar, Maharashtra, India
2Assistant Professor, Rashtriya College of Pharmacy, Hatnoor, Kannad, Chh. Sambhajinagar, Maharashtra, India
Regulatory affairs (RA) professionals play critical roles in the pharmaceutical industry because they are concerned about the healthcare product lifecycle Direction and support for working within rules to speed up the development and delivery of safe and effective healthcare goods to people worldwide, including strategic, tactical, and operational guidance. In pharmaceutical companies, regulatory affairs is a respected, intellectually stimulating, and challenging career.
Regulatory Affairs, also known as government Affairs, is a profession in regulated industries, including pharmaceuticals, clinical gadgets and so on. The Regulatory Affairs profession at its heart is all about gathering, studying and speaking about the danger & amp; advantages of healthcare products to regulatory organizations and the public all globally. Drug Regulatory Affairs is a brand new carrier that has been initiated by governments to defend public health with the say of controlling the production efficacy of products in the area together with prescribed drugs, veterinary drugs treatment, medical devices, insecticides, agrochemicals, cosmetics & amp; complementary medicines. success of regulatory strategies is less dependent on the regulations than on how they are interpreted, applied and communicated within companies and outside constituent. (1)
Importance of Regulatory Affairs
The first point of contact between the corporation and the government agency is the regulatory affairs division. Aids in coordinating research efforts with regulatory requirements. Maximize resource efficiency for the business. Inform the company of the perspective of the government. Impact their companies, strategic decisions. Assist in getting the medication onto the market on time, as even a small delay could negatively impact the company's financial standing. Notify higher authorities with the specific marketing information. .(2)
Need of Regulatory Affairs
Ensuring that their companies namely with all of the system policies and laws pertaining to their business. collaborating with national, state, and local regulatory organizations, including the Food and Drug Administration or the European Medicines Agency. advising their businesses on the regulatory landscapes and issues that could impact suggested initiatives like the support of prescription medication .(3)
Drug regulatory Affair professional
The duty of the Regulatory Affairs expert is to keep up with the continuously changing rules in all of the countries where The business wants to to sell its products. Experts in regulatory affairs are in charge of submitting registration documents to regulatory bodies and conducting all required negotiations to maintain the products' availability on the market. They provide technological and strategic support at the highest levels of their businesses from the outset of a product's development, making a significant financial and clinical contribution to the development effort's advancement and the company's overall success.
The Regulatory Affairs Department's duties are listed below:
COMMON TECHNICAL DOCUMENT (CTD):
A standardized set of standards for the application dossier used to register medicines is known as the Common Technical Document (CTD).It is intended for use in the United States, Europe, and Japan. For the goal of getting ready to submit new medication applications to regional authorities in these countries, the CD format is widely recognized. The Food and Drug Administration (FDA) in the US, the Ministry of Health, Labor, and Welfare in Japan, and the European Medicines Agency (EMA) in Europe worked together to generate this paper. The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) is in charge of maintaining the CTD. (5)
Objectives of CTD
Regulatory Bodies Around Worldwide:

Table No.: 1 Regulatory Bodies Around Worldwide
Modules of CTD
Goals of CTD:
It can be organized into 5 Modules:
Significance
Benefits of CTD
1.Establishing a standard submission format is primarily intended to streamline the application review process and prevent the omission of important information or analysis. If these particulars are omitted, approvals may be unduly delayed.
2. the amount of effort and materials required to to put together applications in order to register human medicines will be significantly reduced by the adoption of a standard layout for technical material. It will also make the process of preparing electronic submissions easier.
3. The utilization of a standardized document containing recurring components will enhance regulatory examinations as well as facilitate communication between the regulatory authorities and the applicant.
4. It is projected that the industry will save a great deal of time and money by using the Common Technical Document (CTD) instead of assembling applications for worldwide registration.
5. Furthermore to raising the bar for Indian norms, the CTD gives the application process broad a methodical framework. The CTD's adoption will also facilitate the ability of regulatory bodies to to communicate information about regulations with one another. (9)
eCTD: Electronic Common Technical Document
The electronic Common Technical Document (eCTD) is used by the pharmaceutical industry to send regulatory data to regulatory agencies. It is based on the Multidisciplinary Group 2 Expert Working Group of the International Conference on Harmonization (ICH) (ICH M2 EWG), which created the Common Technical Document (CTD) format. The eCTD's transport format, which is designed to be easily transferred into the review setting of an agency, environment, makes electronic submissions viable. It facilitates the agency's receipt of regulatory data from the industry and eases the creation, evaluation, life-cycle management, and archiving of electronic submissions. The requirements for guaranteeing the technical correctness of electronic submissions are outlined in the eCTD specifications. This progress in information submission is very helpful to the new drug approval procedure. With a single keystroke in the future, businesses might even be able to submit their applications to several regulatory bodies at once (10)
Requirements to submission of eCTD Documents:
There are two ways For Electronically submission to CDER:
The overview, report body, and individual appendices are its constituent sections.
A top-level folder hierarchy is necessary.
A "backbone" file in Extensible Markup Language (XML) that gives the receiving system lifecycle instructions and metadata about content files.(13)
Hypertext links and bookmarks are techniques used to improve navigation through PDF files. Hypertext links can be identified by blue text or by rectangles with thin lines around them.. The bookmark hierarchy must match the contents table exactly, with no further bookmark levels added on top of what is already there. It is advised to employ no more than four tiers in the hierarchy. When When establishing bookmarks and hyperlinks, make sure to use the Inherit Zoom magnification setting, which will ensure that the destination page appears at the same magnification level as the rest of the content. (14)
File names, including the extension, must not exceed 64 characters. Also, folder names must not exceed 64 characters and the total file folder path length must not exceed 180 characters.
FDA page size for US Letters
All four sides of pages should have a one-inch margin, plus extra space for the binding edge (to prevent problems if documents need to be printed).
Either Times New Roman or Ariel fonts must be used for the content, with body text set at 12 points and tables no less than 10 points.
Agencies should be capable of use Acrobat Reader version 4.0 or higher to read all PDF files. It should not be required for agencies to use any extra software to be able to view and navigate PDF files.
Reusing transferring content from one area to another is made possible by harmonizing standards and all content related to submissions in one location.This is among the greatest ways to accomplish this. This would expedite the submission process by drastically lowering the quantity of repetitive work needed to submit an applicationmany nations or regions. (15)
The publishing workflow shouldn't begin until all source documents have undergone quality control checks.
It is recommended to review all published PDF files on a screen.
Verify the links and bookmarks in published PDF files.
eCTD submissions should always be validated and conformity-checked before being submitted
To submit a sample eCTD, you must first obtain an application number from the Electronic Submissions Team at esub-testing@fda.hhs.gov; this number should only be used for the sample and not for the real eCTD submission. Following submission of our request and contact information, a member of the Electronic Submissions Capability Team will be in contact. and include a Sample Application Number along with additional instructions..
We must obtain a priori issued application number submitting an application in in eCTD configuration to CDER. Pre-assigned application numbers are distinct six-digit numbers, such as 012345, that are given to sponsors so they may be identified
usage. It is required by the FDA to use this number.
whenever you submit an application for eCTD
Advantage of eCTD :
1) advantage over paper submissions is that it is more efficient.
2] Increased Procedure Visibility.
3] Shorter Lead Time and Speedier Approvals.
4] A uniform submission procedure and format.
5] Growth within the marketplace and Revenue.
6] Enhanced Capability to Track.
Publishers may try to prevent validation problems if the annual report is written as a single document by organizing it into a single node that corresponds to one of the annual report sections (e.g., Summary for Nonclinical Studies). This strategy, nevertheless, runs the possibility of creating confusion throughout the review process because the content will not match the node description exactly(16)
NDA APPLICATION
An NDA application must be submitted to the FDA prior to a novel medicine being put on the market. In order to receive this approval, the sponsor provides the NDA A clinical and preclinical examination findings for the analysis of drug information along with a description of the production methods. After the agency receives the NDA, it goes through a technical screening and evaluation process. The evaluation and its supporting documentation ensure that sufficient information and data have been supplied in each area to back up the FDA's official review.
The sponsor may be advised of three possible courses of action once the FDA evaluates an NDA:
Not Approvable: This letter lists the shortcomings and provides an explanation of the decision.
2. Approvable: Thus, it follows that while the drug might be approved, there might be a few minor problems that must be resolved, like labeling changes or a possible need for a commitment to conduct post-approval study.
3. Approval: This signifies that the drug has been given the go-ahead.
Clinical studies are conducted in many phases:
Pre-clinical research: In this study, mice, rats, rabbits, and monkeys are used to evaluate the medication.
Phase IV - post-marketing trial: These studies were carried out after a medication's approval to gather further details.
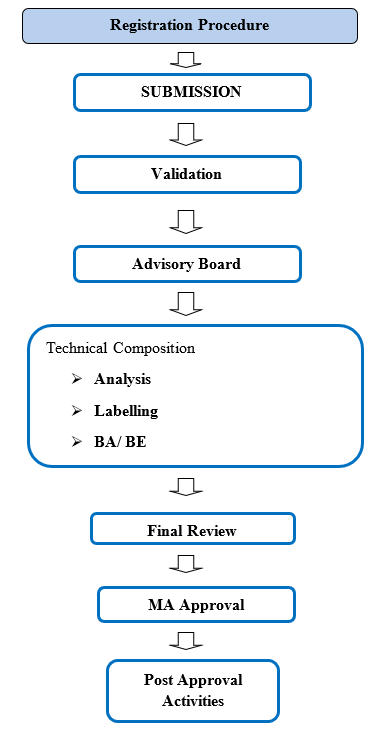
Fig. 1: NDA Approval Procedure
List of New Medicines Authorized for the Year 2023
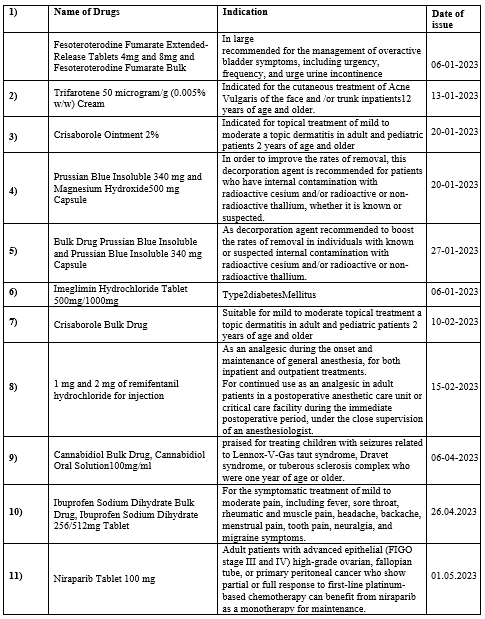
Table No. 2 : List of New Drugs approved in the year 2023
List of New Drugs approved in the year 2024 till date

Table No. 3 : List of New Drugs approved in the year 2024
List Of Pharmaceuticals Prohibited By The Ministry Of Health And Family Welfare From Being Made Or Sold Through Gazettle Notifications Under Section 26a Of The Drugs & Cosmetics Act 1940
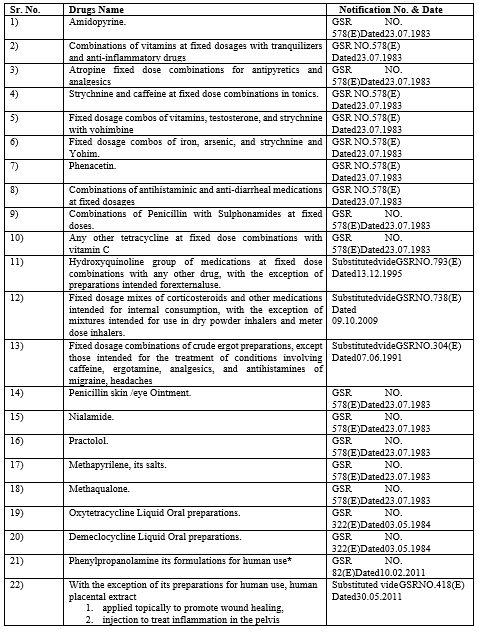
Table No. 4: List of banned Drugs 1940 by the ministry of health and family welfare
*Presently stayed by the Hon’ble High Court of Madras.
**Prohibition was revoked with following conditions vide.
G.S.R.No.367(E)dated13.04.2017:
***The Notification from S.O.Nos 705 (E) to 1048 (E) dated 10.03.2016 were quashed by Hon’ble Delhi High Court vide its order dated 01.12.2016Through the SLP, the Union of India had appealed the Delhi High Court's ruling to the Supreme Court.
REFERENCE


Sanskruti Wagh*, Waghmare S. U., Priyanka Narode, Prasad Shelke, Vaishnavi Borhade, Rohini Kawar, Understanding Regulatory Affairs In The Pharmaceutical Industry: Roles, Importance, And Global Perspectives, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 6, 1052-1067. https://doi.org/10.5281/zenodo.12205641
 10.5281/zenodo.12205641
10.5281/zenodo.12205641