Maharaja Agrasen School of Pharmacy, Maharaja Agrasen University, Baddi
Vascular Cognitive Impairment and Dementia (VCID) is a major, potentially preventable cognitive decline that emerges from cerebrovascular dysfunction, influenced predominantly by aging and metabolic disorders. VCID represents a significant cause of dementia worldwide. VCID arises from heterogeneous cerebrovascular pathologies, including cerebral small and large vessel disease and chronic cerebral hypoperfusion. Therapeutic progress in VCID remains constrained by pathological heterogeneity, frequent overlap with mixed neurodegenerative dementia, limited availability of sensitive early biomarkers, and a persistent shortage of VCID-specific randomized trials validating disease-modifying interventionsThis review summarizes recent diagnostic concepts, mechanistic pathways, and preventive and potential therapeutic interventions in managing VCID. Key mechanisms in pathophysiology of VCID include vascular endothelium disruption and loss of blood–brain barrier integrity, which facilitate oxidative stress and neuroinflammation. These processes impair neurovascular coupling and drive the white-matter injury, disconnecting cortical and subcortical pathways, and subsequent cognitive dysfunction. Hypertension remains the most robust modifiable risk factor, with metabolic disorders, chronic kidney disease, aging, and stroke further accelerating vascular brain injury. Although current management largely focuses on vascular risk reduction, emerging disease-modifying approaches, including senolytics and mitochondrial boosters, necessitate the evaluation in clinical settings. Future progress in VCID management depends on detection of sensitive biomarkers, and phase III clinical trials targeting neurovascular repair.
Dementia affects around 57 million people worldwide as of 2021, with the majority living in low- and middle-income parts of world. The number of dementia cases is predicted to raise to 152 million by 2050, with nearly 10 million new diagnoses are reported each year. Dementia involves a broad spectrum of neurodegenerative and cerebrovascular pathologies, with Alzheimer’s disease being contributing to two-thirds of dementia cases. India alone has 5.3 million dementia patients, and this figure is estimated to double by 2035 due to rapid demographic aging [1][2]. Vascular Cognitive Impairment and Dementia (VCID) is an umbrella term comprising the full specturm of cognitive deficits arising from vascular brain injury, ranging from mild cognitive impairment (mVCI) to established dementia. Within this framework, vascular dementia (VaD) represents the most advanced manifestation of VCID, characterized by cognitive impairment severe enough to compromise daily functioning. Although VCI and VaD share a common vascular etiology, marked by cerebral infarction, hemorrhagic injury, small and large vessel pathology and chronic cerebral hypoperfusion, with difference in clinical severity and functional disability [3]. mVCI is objectively measurable cognitive decline that occurs in the absence of major functional disability and aligns with mild vascular neurocognitive disorder in DSM-5 and mild neurocognitive disorder due to vascular disease in ICD-11[4][5] .Conversely, major VCID, commonly referred to as vascular dementia, comprises conditions such as post-stroke dementia, subcortical ischemic vascular dementia, multi-infarct dementia, and mixed dementia, representing related yet heterogeneous conditions driven by overlapping cerebrovascular and neurodegenerative pathways [6][7] .
Despite recent advances, substantial knowledge gaps persist regarding the early detection of VCID and the development of effective disease-modifying therapies. Recent management of VCID largely relies on managing vascular risk factors and underscoring the urgent need for preventive interventions that can reduce dementia burden, and improve and delay long term cognitive progression.
This review aims to outline vascular dementia and VCID within current DSM-5 and ICD-11 diagnostic frameworks. It explores the underlying cerebrovascular and neuroinflammatory mechanisms driving cognitive impairment. The review also evaluates emerging preventive and therapeutic approaches for managing VCID. Clinical evidence from major intervention trials, including PROGRESS and SPRINT-MIND, are integrated to reform future strategies.
Table 1: Diagnostic framework for Vascular Cognitive Impairment and Dementia across DSM-5 and ICD-11
|
Clinical level |
Diagnostic criteria |
Core clinical description |
Underlying cerebrovascular mechanism |
Corresponding DSM-5/ICD-11 terminology |
|
Non-dementia cognitive impairment |
Mild vascular cognitive impairment |
Cognitive decline detectable only on testing with preserved independence in daily activities |
Chronic cerebral hypoperperfusion |
DSM -5: Mild Vascular Neurocognitive Disorder ICD-11 : Mild Neurocognitive Disorder due to Vascular Disease |
|
Dementia level cognitive impairment |
Post-stroke dementia |
Cognition deterioration emerging with in six months following a stroke |
Cerebral infarcts, large artery cerebrovascular disease |
DSM-5: Major Vascular Neurocognitive Disorder ICD-11: Dementia due to Cerebrovascular disease |
|
|
Subcortical ischemic vascular dementia |
Insidious cognitive decline dominated by executive dysfunction and psychomotor slowing |
Cerebral small vessel disease with lacunes and white matter lesions |
DSM-5: Major Vascular Neurocognitive Disorder ICD-11: Dementia due to Cerebral Small Vessel Disease |
|
|
Multi-infarct dementia |
Progressive cognitive impairment linked to recurrent infarctions |
Multiple cortical infarcts related to large vessel pathology |
DSM-5: Major Vascular Neurocognitive Disorder ICD-11: Dementia due to Cerebrovascular Disease |
|
|
Mixed dementia |
Cognitive impairment attributable to combined vascular and neurodegenerative pathology |
Coexisting cerebrovascular and Alzheimer’s like changes |
DSM-5: Major Neurocognitive Disorder due to multiple etiologies ICD-11 Mixed Dementia |
MECHANISTIC BASIS OF VCID
Cerebral small artery disease
Cerebral small vessel disease (CSVD) is a key determinant in pathogenesis of VCID, particularly subcortical ischemic vascular dementia [8]. Degenerative changes in cerebral blood vessels such as arteriolosclerosis and lipohyalinosis causes endothelial dysfunction in small penetrating arterioles and capillaries, compromise cerebral blood flow and neurovascular adaptability [9]. These alterations result in chronic cerebral hypoperfusion and increased susceptibility to ischemic injury within subcortical and deep white-matter regions [10]. These cumulative effects lead to disconnection of fronto-subcortical circuits, manifesting clinically as impairment in executive and cognitive dysfunction [11].
Large vessel disease and Infarction
Cerebral large vessel disease (CLVD) contributes to vascular cognitive impairment through atherosclerotic stenosis and embolic occlusion of major cerebral arteries, culminating in cortical and subcortical infarctions [12]. These vascular events cause abrupt reduction in regional cerebral perfusion and metabolic failure causing irreversible cortical and subcortical neuronal loss [10]. Infarctions in cortical and subcortical regions disrupt connectivity across executive, and memory neural pathways [13]. In addition to focal tissue damage, large vessel infarction initiates secondary processes such as Wallerian degeneration and transneuronal diaschisis which further amplify cognitive deficits [14]. The cummulative burden of recurrent and multifocal infarcts, specifically in the presence of coexisting small vessel disease, produces a trajectory of cognitive decline and accelerates progression to vascular dementia.
Chronic Hypoperfusion
Chronic cerebral hypoperfusion induces cognitive decline through persistent impairment of neurovascular homeostasis rather than focal ischemic injury. Prolonged reductions in cerebral blood flow compromise oxygen and glucose delivery, leading to impaired mitochondrial respiration and subsequent metabolic energy failure, and increased production of reactive oxygen species within neurons and glia [15]. Hypoperfusion induced oligodendrocyte injury and subsequent axonal degeneration facilitate the breakdown of essential cortical and subcortical pathways, serving as the significant driver for slowed processing speed and executive dysfunction. [16]. In parallel to chronic hypoperfusion, disruption of the blood–brain barrier promotes neuroinflammation and the buildup of toxic metabolites, further exacerbate synaptic dysfunction and neuronal loss [17]. These interrelated pathways cognitive decline and potentiate the effects of coexisting infarction towards the VCID advancement.
Blood brain barrier dysfunction
Dysfunction of the blood–brain barrier (BBB) represents a primary mechanism connecting vascular pathology in the brain to cognitive decline. When BBB integrity is compromised, it allows abnormal bidirectional movement of molecules between bloodstream and brain tissue. BBB breakdown arises from diverse contributing factors such as damage to endothelial cells lining cerebral blood vessels, loss of supportive pericytes, and degradation of tight junction proteins that maintain BBB integrity [18]. These pathological changes in BBB results in the leakage of plasma proteins such as fibrinogen and pro-inflammatory molecules into the brain interstitial space [19]. The entry of plasma-derived proteins and inflammatory molecules into interstitial space activates a series of pathogenic responses that exacerbate cerebral injury. Microglia, the brain's resident immune cells, become activated, while astrocytes enter a reactive state. Together, these processes disrupt the delicate signaling between neurons at synapses and threaten neuronal survival [20]
Neuroinflammation and Oxidative stress
Reactive oxygen species (ROS) are significant drivers of cognitive decline in cerebrovascular disease. Vascular injury, chronic hypoperfusion and BBB dysfunction activate microglia and astrocytes, leading to sustained release of pro-inflammatory cytokines and reactive oxygen and nitrogen species [21]. Furthermore, ROS and proinflammatory cytokines together impair synaptic transmission by disrupting glutamate clearance, altering postsynaptic glutamate receptor function, and destabilizing calcium homeostasis [22]. These events together suppresses synaptic plasticity and long term potentiation mediated by glutamate and exaggerate synaptic loss [23]. Oxidative stress damages mitochondrial proteins and mitochondrial DNA, causes inhibition of oxidative phosphorylation, and subsequent decrease in ATP production. Mitochondrial bioenergetics failure at synapses induces diminished neurotransmitter release, further weakening neuronal communication [24]. Oxidative injury also affects oligodendrocytes, promoting demyelination and axonal dysfunction within cortical and subcortical neural pathways. Long standing neuroinflammation accelerate the advancement of VCID [25
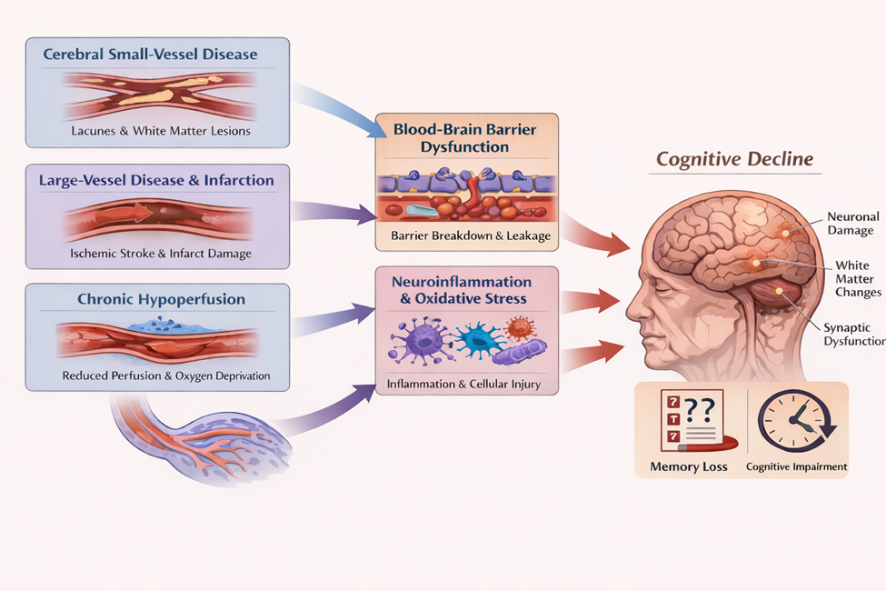
Fig 1. Pathophysiological changes in VCID
DETERMINANTS OF VCID
1. Hypertension
Hypertension is a major preventable risk factor for VCID, as it induces sustained structural and functional damage to small and large cerebral blood vessels. Several prospective cohort and longitudinal studies indicate that hypertension is associated with a significant, 59 % increased risk of vascular dementia, compared with normotensives counterparts[26]. Persistent elevation of blood pressure promotes arteriolosclerosis and lipohyalinosis of small penetrating arteries, impair cerebral vessel autoregulation and decreases perfusion to subcortical and deeper white matter regions [27]. At the same time, hypertension accelerates atherosclerosis in the major cerebral arteries, heightening the incidence of cortical infarcts and the consequent stroke that impair cognition [28]. Hypertension induced mechanical stress on cerebral vessels disrupts endothelial signaling, causes impaired neurovascular coupling and loss of BBB integrity, thereby accelerating progression across the VCID spectrum [29]
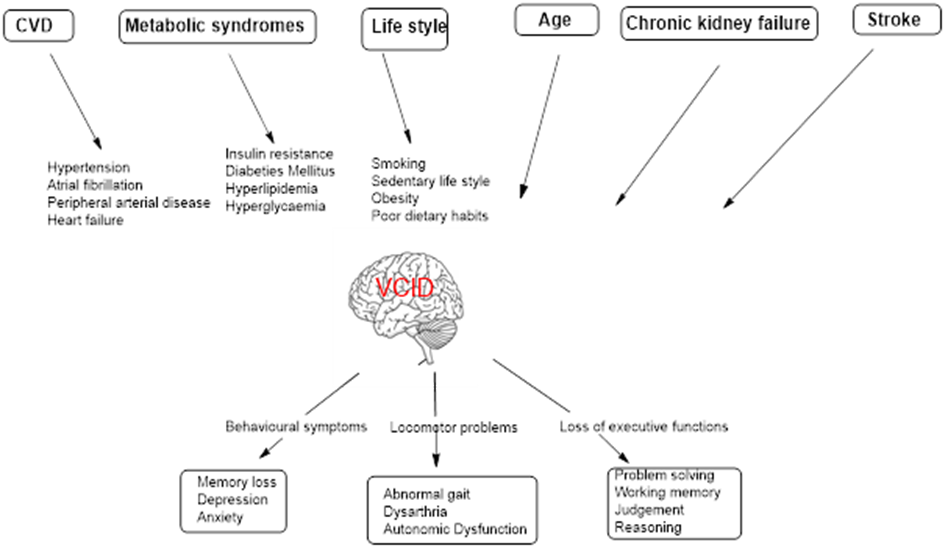
Fig 2. Manifestations of VCID
2. Metabolic syndromes
Metabolic disorders including hyperlipidemia, insulin resistance and diabetes mellitus contribute to VCID through convergent vascular and cellular mechanisms. Hyperlipidemia accelerates atherosclerosis in large and medium cerebral arteries and promote lipid-mediated endothelial inflammation. Chronic hypoperfusion by the cerebral arteries increases the risk of cortical infarction. Elevated circulating low-density lipoprotein (LDL), penetrates the arterial intima and undergoes oxidative changes, triggers endothelial dysfunction and inflammatory cell recruitment. Macrophage uptake of oxidized LDL leads to foam cell formation and fatty streak development, while sustained lipid-driven inflammation promotes plaque formation and progressive luminal narrowing [30].
Insulin resistance and diabetes mellitus expose cerebral microvessels to hyperglycemia, dyslipidemia, and oxidative stress. This carbohydrate and lipid mediated injury stimulates excess extracellular matrix deposition, leading to basement membrane thickening in the endothelial cells and reduced capillary flexibility .Endothelial dysfunction further drive capillary rarefaction and impaired NO mediated vasodilation [31].Advanced glycation end-product (AGEs) accumulate in cerebral blood vessels and activate RAGE (Receptors for Advanced Glycation End products) mediated endothelial dysfunction [32]. These metabolic insults s amplify neuroinflammation, white-matter degeneration, and synaptic dysfunction in the cortical regions of brain, driving advancement from mild vascular cognitive deficits to established dementia [33].
3. Life Style
A sedentary lifestyle reduces skeletal muscle glucose uptake by decreasing insulin-stimulated GLUT4 translocation and mitochondrial antioxidant defense, leading to inefficient glucose utilization and impaired mitochondrial bioenergetics [34]. Physical inactivity also lowers muscle mass by decreasing muscle protein synthesis and activating proteolytic system. Concurrently, physical inactivity promotes lipid accumulation with in the skeletal muscle, generate lipotoxic intermediates such as DAG, ceramides, that activate serine kinases and impair insulin signaling [35]. Obesity aggravates insulin resistance by expanding adipose tissue, which releases excess free fatty acids into the circulation and drives ectopic lipid accumulation in liver and skeletal muscle [36]. These peripheral metabolic disturbances reduce endothelial nitric oxide signaling [37], disrupt neurovascular coupling[38], and promote cerebral microvascular remodeling, culminating in chronic cerebral hypoperfusion and BBB breakdown [39]. The resulting white-matter injury and synaptic energy failure weaken cognitive pathways, thereby contributing towards VCID continuum [40].
4. Age
Ageing promotes VCID through progressive structural and functional deterioration of the cerebral vasculature and neurovascular unit. With advancing age, cerebral arteries and arterioles undergo increased stiffness, collagen deposition, and loss of elastin, causing reduced vascular elasticity and impaired regulation of cerebral blood flow [41]. Age-related endothelial dysfunction diminishes nitric oxide bioavailability and further limits cerebral blood flow during cognitive demand [42].Aging disrupts BBB integrity through oxidative stress, senescence of vascular cells, and neurovascular unit instability, leading to enhanced permeability of BBB.
Inflammatory ROS signaling promotes degradation of tight junction proteins such as occludin,. Claudins and ZO-1, and consequent increase in paracellular leakage. Simultaneously, pericyte loss and astrocyte end feet deattachment further weaken the BBB support [43]. These vascular alterations promote chronic cerebral hypoperfusion, white-matter degeneration, and microinfarcts , which preferentially disrupt subcortical and cortical neural pathways, critical for cognition [44]. Together, age related vascular and neurovascular changes contribute towards the advancement of VCID.
5. Chronic kidney failure
Chronic kidney failure (CKD) contributes to VCID through tightly linked vascular, metabolic, and inflammatory mechanisms. Accumulation of uremic toxins in reduced renal clearance (e.g., indoxyl sulfate, p-cresyl sulfate) induce systemic endothelial dysfunction and impair cerebral autoregulation [45]. This disrupts tight junction proteins, increasing BBB permeability and allowing inflammatory mediators to enter the brain parenchyma. The ensuing microglial activation and neuroinflammatory signaling preferentially damage oligodendrocytes and axonal tracts. These processes accelerate white-matter injury and compromise subcortical neurons, promoting progression of VCID [46]. Collectively, CKD driven vascular toxicity and systemic metabolic comorbidities converge to aggravate cerebral small vessel dysfunction, BBB breakdown, and neuroinflammatory white-matter degeneration, thereby accelerating the trajectory of VCID.
6. Stroke
Stroke induces VCID through an acute ischemic cascade of events followed by progressive cerebrovascular pathology. Diminished cerebral blood flow rapidly depletes ATP, disabling the energy-dependent Na?/K?-ATPase and collapsing transmembrane ion gradients. Loss of ionic homeostasis causes sustained membrane depolarization and reversal of glutamate transporters, while depolarization-induced vesicular release further elevates extracellular glutamate. Excess glutamate overactivates NMDA and AMPA receptors, driving pathological events [47]. In the peri ischemic region, partial energy failure activates mitochondrial apoptosis and death receptor signaling, resulting in delayed neuronal loss, specifically when lesions involve strategic cognitive regions such as the thalamus, frontal cortex, or limbic pathways. Long-term cognitive decline develops from the cumulative burden of recurrent cerebral infarction and progressive small vessel pathology. Together, these excitotoxic and apoptotic processes, aggravate vascular remodeling and disconnected neural pathways, ultimately drive progressive evolution from poststroke cognitive impairment to established vascular dementia [48].
PREVENTIVE INTERVENTIONS FOR VASCULAR COGNITIVE IMPAIRMENT AND DEMENTIA
VCID might be prevented through two types of interventions. The first one is an early identification and medical treatment of cardiovascular conditions. An effective control of cardiovascular risk factors prevents VCID and may be more effective than therapeutic Intervetions [49]. Hypertension is associated with leukoaraiosis, or lesions in periventricular and subcortical white matter regions of brain [50]. Studies suggest that preserving ideal cardiovascular health in middle age contributes to better cognitive outcomes in older adulthood [51]. Another potentially important preventative intervention against VCID is to build neurocognitive reserve. The concept of neurocognitive reserve describes the ability to compensate age related brain pathology [52]. Neurocognitive reserve includes 1) Cerebral reserve related to structural differences in brain architechture in terms of neuronal and synaptic density [53]; 2) Cognitive reserve is about differences in ability to make efficient use of existing brain resources when engaged in cognitive task [54]; 3) Neural network maintenance is the differential capacity to repair impaired synapses, synaptogenesis, and neurogenesis [55]. Citicoline, an essential intermediate in the biosynthesis of structural phospholipids in cell membranes, has been reported to exhibit neuroprotective activity. Citicoline may provide neuroprotective benefits in VCID by enhancing phospholipid membrane repair, supporting cholinergic neurotransmission, and reducing oxidative stress. [56][57] , although clinical benefits in ischemic stroke induced VCID, remains unexplored [58]. With growing insights into cognitive processes, boosting neurocognitive reserve through pharmacological strategies may emerge as a future therapeutic approach. Caution, however , needs to be taken, as the concept of neurocognition reserve implies both the chance to mitigate VCID by increasing the cerebral reserve and the danger of underestimating severity of cerebrovascular damage when just evaluating the VCID [52].
Collectively, evidence from observational studies, randomized controlled trials, and meta-analyses indicates that hypertension is a major modifiable driver of VCID. Randomized evidence from trials including SPRINT-MIND and PRESERVE supports the cognitive relevance of intensive blood pressure lowering, even in older populations.
These interventions consistently slow white-matter hyperintensity (WMH) progression without demonstrating adverse effects on cerebral perfusion [59]. More recent analyses suggest that calcium channel blockers and angiotensin II receptor blockers may offer additional protection against dementia beyond BP lowering alone [60]. Secondary analyses further indicate that angiotensin-converting enzyme inhibitors and calcium channel blockers may preferentially attenuate WMH progression [61]. Despite this, short term clinical investigations report no differential effects on cerebrovascular rreserve [62]. Taken together, the data suggest hypertension as a key modifiable determinant of VCID, while underscoring the need for adequately long duration clinical trials to determine whether antihypertensive agents provide cognitive protection beyond blood pressure lowering effects.
Beneficial effects of changing life styles and physical activity (diet, exercise, and environmental enrichment) on VCID and VaD have been reported [63][64]. In rats with 2-VO induced chronic cerebral hypoperfusion, treadmill exercise for 4 weeks has been found to reduce cognitive decline by enhancing neurogenesis mediated by upregulation of BDNF expression [64]
Although elevated cholesterol has been implicated in cerebrovascular dysfunction and may contribute to VCID progression, the therapeutic effect of cholesterol reduction in VCID and VaD is yet to be determined. A recent Cochrane Database Review has indicated that statins do not prevent cognitive decline when given in late life to the high risk people for vascular disease [65]. Clinically, antidiabetic therapy particularly metformin and long-term statins were able to demonstrate a modest reduction in cognitive decline and dementia risk, especially among individuals with high vascular burden [66][67]. Clinical trials with improvement in cognition as the main outcome, show variable results, while VCID-specific randomized studies are comparatively lacking. [68].
CKD is increasingly recognized as a systemic vascular brain disorder, contributing to VCID. Large cohort studies show a graded increase in dementia risk with declining endothelial glomerular filtration rate (eGFR), independent of traditional vascular risk factors.[69]. ACE inhibitors and angiotensin receptor blockers (ARBs) mitigate Renin Angiotensin Aldosterone System (RAAS) driven endothelial dysfunction, microvascular injury, and resulting hypertension in CKD, which may moderately delay cognitive decline. In parallel, Sodium Glucose Cotransporter 2 (SGLT2 ) inhibitors provide renal protection by improving glomerular filtration rate, reducing albuminuria, and suppressing inflammatory and oxidative stress pathways, thereby delaying CKD advancement. Cardiovascular benefits of SGL2 and associated reductions in stroke risk may further contribute to cognitive preservation [70].
Age is the dominant non-modifiable risk factor for VCID, acting through vascular stiffening, impaired autoregulation and endothelial senescence induced decreased nitric oxide signaling. Importantly, aging magnifies the deleterious vascular insults associated with disorders including stroke and CKD. Rapamycin and metformin has shown anti-aging neurovascular benefits in preclinical studies by improving cerebral perfusion, preservation of BBB integrity, and cognition [77]. Furthurmore, epidemiological evidence links metformin use with reduced cognitive decline and dementia risk, supporting potential translational relevance [70] [71][72]. Senolytics are the drugs that selectively eliminate senescent cells, that have permanently stopped dividing, and accumulated with age. Preclinical studies have demonstrated that targeting senescent cerebrovascular cells, improve BBB integrity, reduce neuroinflammation, and enhance cognitive performance in aged animal models highlighting potential vascular benefits of senolytic therapy in aging brain [73]. While preclinical evidence supports the neuroprotective role of senolytics, their therapeutic relevance in VCID remains untested in human trials. NAD? is Nicotinamide Adenine Dinucleotide (NAD?), is a vital cellular mitochondrial bioenergetics regulator. NAD? supplementation supports mitochondrial energetics and endothelial nitric oxide signaling in a rodent model of ageing, thereby restoring neurovascular coupling and improving vasodilatory function. These vascular and mitochondrial improvements are associated with better cognitive and motor coordination in preclinical aging models, although human cognitive evidence remains inconclusive [73] [74].
Stroke is the strongest clinical determinant of VCID with both symptomatic and silent infarcts causes irreversible neural network disconnection, chronic neuroinflammation, BBB integrity, and acceleration of cerebral small vessel disease. Experimental models provide evidence that ischemia initiates neuroinflammation induced white-matter degenerative processes that drive later cognitive dysfunction [75]. In parallel, clinical data demonstrate that post-stroke VCID occurs in 30–50% of stroke survivors, with risk amplified by recurrent strokes [76] . Supporting a preventative strategy, randomized evidence shows that antihypertensive and statin therapy can mitigate VCID development by limiting recurrent vascular injury and slowing cSVD progression. [77]. There is no FDA approved specific for post stroke VCID. Blood pressure management is the strongest pharmacological prevention strategy for stroke-induced VCID.A meta-analysis suggests that memantidine is promising treatment for post stroke induced vascular cognitive impairment [78].
DISCUSSION
Overall, recent evidences supports risk modifying rather than disease modifying interventions with cognitive benefits, underscoring the need for VCID-focused trials. While preclinical evidence supports the neuroprotective role of senolytics, their therapeutic relevance in VCID remains unexplored in human clinical studies. Despite strong preclinical evidence for improved mitochondrial function with NAD? repletion, human cognitive benefits remain inconclusive and require further investigation. Hypertension promotes cerebrovascular injury through endothelial dysfunction and increases risk of clinical and covert stroke. Landmark clinical trials such as PROGRESS demonstrate that antihypertensive therapy reduces recurrent stroke and consequent cognitive decline, indicating that stroke prevention is a central mechanism by which blood pressure control mitigates vascular cognitive impairment. SPRINT-MIND study provides additional evidence that intensive blood pressure lowering lowers mild cognitive impairment risk, supporting the role of hypertension-driven neurovascular dysfunction as a key pathway linking stroke to dementia.
Blood pressure management represents one of the strongest modifiable strategies for preventing and slowing VCID progression. In PROGRESS, perindopril based therapy after stroke or transient ischemic attack reduced cognitive decline and dementia linked to recurrent cerebrovascular events, effective for secondary prevention in high-risk populations [79]. Extending this evidence, SPRINT-MIND showed that intensive systolic BP control (<120 mmHg) significantly lowered mild cognitive impairment and effective for primary prevention in hypertensive adults, with sustained benefit confirmed in long-term follow-up analyses through seven years [59]. The China Rural Hypertension Control Project (CRHCP), a large-scale cluster-randomized trial involving over 30,000 participants, demonstrated that blood pressure control intervention significantly reduced the risk of dementia by 15% and cognitive impairment by 16% [80]. The SPRINT-MIND provided the proof in a highly controlled environment, this Rural China study provided the "real-world relevance" in a general population with fewer medical resources. Mechanistic MRI substudies reinforce these clinical findings by showing attenuation of white-matter lesion progression and preservation of cerebral perfusion with intensive therapy, consistent with reduced small-vessel disease burden [59][81]. Collective real world evidences supports early and stable BP control as a cornerstone intervention for reducing VCID and vascular dementia [82].
CONCLUSION
VCID considerably impacts social life by impairing executive functions such as planning and communication, leads to social withdrawal and loss of independence. Primary risk factors are vascular including hypertension and stroke. Preventive strategies for VCID focuses on “what is good for heart is good for brain”, specifically intensive blood pressure control and regular exercise.There is an urgent need for clinical trial to approve new drugs for VCID.
REFERENCES

Gaganjit Kaur, Vascular Cognitive Impairment and Dementia : From Pathophysiological Mechanisms to Emerging Therapeutic Strategies, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 323-340. https://doi.org/10.5281/zenodo.18871014
 10.5281/zenodo.18871014
10.5281/zenodo.18871014
