
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
12Chemists College of Pharmaceutical Sciences and Research, Kochi-Kerala
3Bachelor of Siddha Medicine and Surgery, Sri Sai Ram Siddha Medical College
Background: Peptide therapeutics occupy a unique regulatory intersection between small molecules and biologics, creating jurisdictional classification challenges. While the U.S. Food and Drug Administration (FDA) and India's Central Drugs Standard Control Organization (CDSCO) both regulate these modalities, significant heterogeneity exists in approval pathways, Chemistry Manufacturing and Controls (CMC) requirements, and clinical trial frameworks. Objectives: This review systematically compares regulatory requirements for synthetic and recombinant peptide therapeutics between the FDA and CDSCO, identifying critical gaps, convergence opportunities, and strategic considerations for multi-regional drug development. Methods: A comprehensive narrative review of regulatory guidelines, pharmacopeial standards, and legislative frameworks was conducted. Databases including PubMed, Scopus, and regulatory repositories (FDA.gov, CDSCO.gov.in) were searched for documents published between 2010–2024 using keywords: "peptide therapeutics," "regulatory approval," "FDA," "CDSCO," "biosimilars," and "CMC requirements." Primary regulatory guidance documents, ICH guidelines, and official gazette notifications were analyzed. Key Findings: Classification thresholds differ substantially: FDA categorizes peptides <40 amino acids synthesized chemically as small molecules (505(b)(2) pathway), whereas CDSCO applies biologic-like scrutiny based on manufacturing complexity regardless of size. CMC requirements for impurity profiling (elemental impurities, residual solvents) and immunogenicity risk assessment show partial harmonization with ICH Q6B and S6(R1), but divergence persists in post-approval change management and biosimilar extrapolation criteria. Conclusion: While both jurisdictions increasingly align with ICH standards, peptide-specific regulatory science remains fragmented. Strategic development programs must navigate divergent classification logic, particularly for complex peptides and biosimilar products, necessitating early regulatory intelligence and QbD (Quality by Design) implementation
Peptide therapeutics have emerged as a rapidly expanding class of pharmaceutical agents, occupying a distinctive position at the interface of small-molecule drugs and biologics. Structurally composed of short chains of amino acids, peptides exhibit high target specificity, favorable safety profiles, and predictable metabolic pathways, making them particularly attractive for the treatment of metabolic disorders, oncology, infectious diseases, cardiovascular conditions, and rare genetic disorders.[1] Over the past two decades, advances in solid-phase peptide synthesis (SPPS), recombinant DNA technology, and analytical characterization have substantially improved the feasibility of large-scale peptide manufacturing, thereby accelerating clinical translation and commercial development. Despite their therapeutic promise, peptide drugs present unique regulatory challenges due to their intermediate molecular complexity, susceptibility to degradation, potential immunogenicity, and sensitivity to manufacturing variations.[2] Unlike conventional small molecules, peptides often exhibit structural heterogeneity arising from sequence truncations, racemization, aggregation, and post-synthetic modifications. Conversely, although peptides share certain attributes with biologics, many lack the higher-order structural complexity typically associated with proteins, complicating their regulatory classification. As a result, peptide therapeutics are subject to heterogeneous regulatory treatment across global jurisdictions, particularly with respect to product classification, Chemistry, Manufacturing, and Controls (CMC) requirements, clinical development expectations, and post-approval lifecycle management.[3] In the United States, peptide therapeutics are regulated by the Food and Drug Administration (FDA) under multiple statutory frameworks, primarily the Federal Food, Drug, and Cosmetic Act (FD&C Act) and, in select cases, the Public Health Service (PHS) Act. The FDA generally classifies chemically synthesized peptides particularly those comprising fewer than 40 amino acids as small-molecule drugs, thereby permitting approval through New Drug Application (NDA) pathways such as 505(b)(1) or 505(b)(2).[4] In contrast, recombinant peptides and complex synthetic constructs may be regulated under biologics-oriented paradigms, necessitating more stringent controls on manufacturing consistency, comparability, and immunogenicity risk assessment. This nuanced classification framework, while scientifically grounded, introduces regulatory ambiguity for developers of novel or hybrid peptide modalities. In India, peptide therapeutics fall under the regulatory purview of the Central Drugs Standard Control Organization (CDSCO), operating through the Drugs and Cosmetics Act, 1940 and associated rules.[5] Unlike the FDA’s size-based classification logic, CDSCO adopts a more process-centric and risk-based approach, often subjecting peptide products regardless of molecular length to regulatory scrutiny comparable to that applied to biologics. This includes heightened expectations for preclinical safety evaluation, comprehensive CMC documentation, and, in certain cases, local clinical trial requirements. The absence of peptide-specific regulatory guidance in India further amplifies interpretative variability, increasing development complexity for both domestic manufacturers and multinational sponsors.[6]
Harmonization efforts led by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) have partially addressed these challenges by establishing globally accepted standards for quality, safety, and efficacy. Guidelines such as ICH Q6B (Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products), ICH S6(R1) (Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals), and ICH Q8–Q12 (Quality by Design and lifecycle management) provide a foundational framework for peptide drug development.[7] However, the application of these guidelines to peptides remains inconsistent, particularly in areas such as impurity profiling, immunogenicity assessment, biosimilar development, and post-approval change management. Given the growing clinical and commercial importance of peptide therapeutics, a systematic and comparative evaluation of regulatory requirements across major jurisdictions is urgently needed.[8] Understanding regulatory divergence and convergence between the FDA and CDSCO is essential for optimizing global development strategies, minimizing regulatory risk, and facilitating timely patient access to innovative peptide-based therapies. This review therefore aims to critically analyze and compare the regulatory frameworks governing peptide therapeutics in the United States and India, with a specific focus on classification criteria, preclinical and clinical requirements, CMC expectations, marketing authorization pathways, and post-marketing obligations. By identifying regulatory gaps and alignment opportunities, this work seeks to inform evidence-based regulatory decision-making and support the rational design of multi-regional peptide development programs.[9]
2. OVERVIEW OF PEPTIDE THERAPEUTICS
Peptide therapeutics constitute a structurally and functionally diverse class of medicinal products characterized by amino acid sequences typically ranging from two to approximately fifty residues. Positioned between conventional small-molecule drugs and large protein biologics, peptides combine the pharmacological precision of biologics with the synthetic accessibility of small molecules.[10] Their mechanism of action is predominantly mediated through high-affinity interactions with endogenous receptors, enzymes, ion channels, and signaling proteins, enabling modulation of complex biological pathways with minimal off-target effects. From a pharmacodynamic perspective, peptides demonstrate superior selectivity and potency compared to small molecules, owing to their ability to mimic endogenous ligands and participate in highly specific molecular recognition processes.[11]
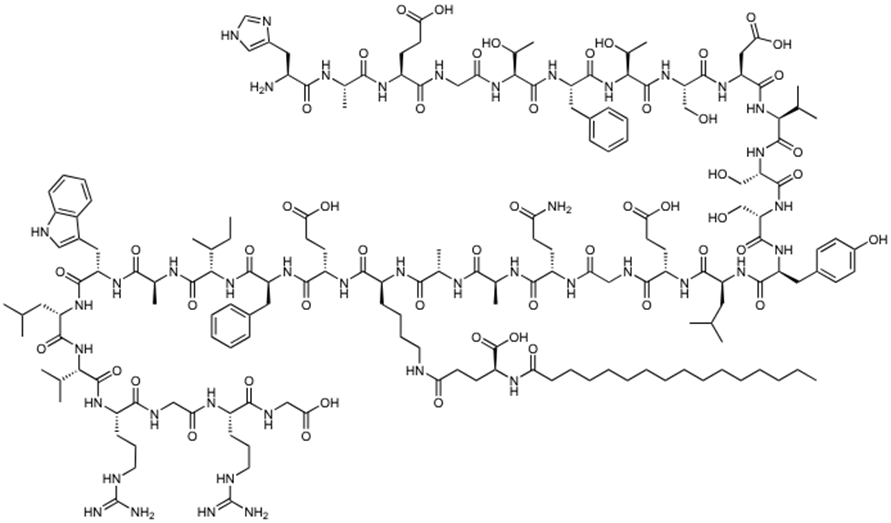
Figure 1. GLP-1 receptor agonists (Semaglutide).
This intrinsic specificity often translates into improved safety margins and reduced systemic toxicity. However, their pharmacokinetic profiles are frequently limited by rapid enzymatic degradation, low oral bioavailability, and short systemic half-lives, necessitating specialized formulation strategies and alternative routes of administration, including parenteral, transdermal, and intranasal delivery systems.[12] Peptide therapeutics can be broadly classified based on their origin and manufacturing approach into chemically synthesized peptides, recombinant peptides, and complex peptide conjugates. Chemically synthesized peptides, commonly produced using solid-phase peptide synthesis (SPPS), offer precise sequence control and scalability for short to medium-length peptides.[13] Recombinant peptides, produced via microbial or mammalian expression systems, are generally employed for longer or structurally complex peptides requiring post-translational modifications. Hybrid modalities, including peptide–drug conjugates, lipidated peptides, and PEGylated peptides, have further expanded the therapeutic landscape by enhancing stability, bioavailability, and target engagement. Manufacturing processes play a critical role in defining the regulatory and quality attributes of peptide drugs.[14] Peptide synthesis is inherently susceptible to process-related impurities such as deletion sequences, epimerization, oxidation, deamidation, residual protecting groups, and inorganic contaminants. Even minor variations in synthesis conditions, purification strategies, or scale-up parameters can significantly impact product quality, immunogenicity risk, and clinical performance. Consequently, robust analytical characterization using orthogonal techniques including high-resolution mass spectrometry, chromatographic profiling, peptide mapping, and structural conformation analysis is essential to establish identity, purity, and batch-to-batch consistency.[15] Clinically, peptide therapeutics have demonstrated substantial success across multiple therapeutic areas. Notable examples include glucagon-like peptide-1 (GLP-1) receptor agonists for diabetes and obesity, gonadotropin-releasing hormone (GnRH) analogs for endocrine disorders and oncology, somatostatin analogs for neuroendocrine tumors, and antimicrobial peptides targeting resistant pathogens. The expanding pipeline of peptide-based drugs underscores their growing clinical relevance and commercial viability, with several products achieving blockbuster status in global markets. Despite these advantages, peptide therapeutics pose distinct regulatory challenges arising from their dual identity as chemically defined yet biologically active molecules.[16] Regulatory authorities must balance traditional small-molecule evaluation paradigms with biologics-oriented considerations, particularly in areas such as immunogenicity assessment, comparability, and biosimilar development. The absence of universally harmonized peptide-specific regulatory guidelines further complicates product development, especially for sponsors pursuing simultaneous or sequential approvals across multiple jurisdictions. In this context, a clear understanding of the scientific and technological attributes of peptide therapeutics is fundamental to interpreting regulatory expectations.[17] The physicochemical properties, manufacturing complexity, and clinical behavior of peptides directly influence regulatory classification, preclinical safety requirements, CMC documentation, and lifecycle management strategies. This section therefore provides the foundational framework necessary to contextualize the subsequent comparative analysis of regulatory pathways governing peptide therapeutics in the United States and India.[18]
Table 1. Classification of peptide therapeutics based on origin and structural complexity. [19]
|
Classification basis |
Type of peptide |
Description |
Representative examples |
Regulatory relevance |
|
Manufacturing origin |
Chemically synthesized peptides |
Short to medium-length peptides produced via solid-phase peptide synthesis (SPPS) |
GnRH analogs, somatostatin analogs |
Often regulated as small molecules |
|
Recombinant peptides |
Peptides produced using microbial or mammalian expression systems |
Insulin, growth hormone fragments |
Subject to biologics-like scrutiny |
|
|
Structural modification |
Native peptides |
Unmodified sequences identical or similar to endogenous peptides |
Glucagon |
Lower immunogenic risk |
|
Modified peptides |
Chemically altered to enhance stability or potency |
Liraglutide, semaglutide |
Increased CMC and safety requirements |
|
|
Conjugation status |
Peptide conjugates |
Peptides linked to drugs, lipids, or polymers |
Peptide–drug conjugates |
Hybrid regulatory evaluation |
3. REGULATORY FRAMEWORK IN THE USA
3.1 Regulatory Authority
In the United States, peptide therapeutics are regulated by the U.S. Food and Drug Administration (FDA), primarily through the Center for Drug Evaluation and Research (CDER). Regulatory oversight is governed by the Federal Food, Drug, and Cosmetic Act (FD&C Act), with additional statutory considerations under the Public Health Service (PHS) Act for select biologic products. The FDA adopts a science- and risk-based regulatory approach, wherein peptide classification and approval pathways are determined by molecular characteristics, manufacturing processes, and clinical risk profiles rather than therapeutic intent alone. Unlike traditional biologics regulated under section 351 of the PHS Act, most peptide therapeutics fall within the scope of drugs regulated under section 505 of the FD&C Act. However, recombinant peptides, fusion constructs, and highly complex peptide modalities may be evaluated using regulatory principles aligned with biologics, particularly in the context of comparability, immunogenicity, and manufacturing change control. This hybrid regulatory treatment reflects the intermediate nature of peptides and underscores the FDA’s flexible, case-by-case assessment paradigm.[20]
3.2 Regulatory Guidelines for Peptides
The FDA does not currently issue peptide-specific standalone regulatory guidance; instead, peptide therapeutics are evaluated using a combination of small-molecule and biologics regulatory frameworks. Chemically synthesized peptides, particularly those comprising fewer than 40 amino acids, are generally regulated as small-molecule drugs, while longer or structurally complex peptides may be subject to additional biologics-oriented requirements. Key FDA guidance documents applicable to peptide therapeutics include those addressing nonclinical safety evaluation, impurity profiling, pharmaceutical development, and immunogenicity risk assessment. International Council for Harmonisation (ICH) guidelines adopted by the FDA such as ICH Q6A/Q6B (Specifications), ICH S6(R1) (Preclinical Safety Evaluation), ICH Q8–Q11 (Quality by Design), and ICH Q12 (Lifecycle Management) form the backbone of regulatory expectations for peptide development. The absence of peptide-specific regulatory guidance, however, necessitates proactive regulatory engagement and scientific justification by sponsors.[21]
3.3 Preclinical Requirements
Preclinical evaluation of peptide therapeutics in the United States is conducted in accordance with FDA guidance and ICH S-series guidelines. Toxicological assessment typically includes acute, subacute, and chronic toxicity studies, with study design tailored to the intended clinical indication, dosing duration, and route of administration. For peptides closely resembling endogenous molecules, the FDA may permit a reduced nonclinical package, provided adequate scientific justification is presented. Immunogenicity assessment represents a critical component of peptide preclinical development. Although peptides generally exhibit lower immunogenic potential than large biologics, sequence modifications, aggregation, impurities, and formulation components may elicit immune responses. The FDA expects a risk-based immunogenicity evaluation strategy incorporating in silico, in vitro, and in vivo assessments, particularly for novel peptide entities or long-term administration.[22]
3.4 Clinical Trial Design and Requirements
Clinical development of peptide therapeutics in the United States proceeds under an Investigational New Drug (IND) application. Phase I studies typically focus on safety, tolerability, pharmacokinetics, and pharmacodynamics, with adaptive designs increasingly employed for dose optimization. Due to their high target specificity, peptides often demonstrate predictable dose–response relationships, facilitating efficient early-phase development. The FDA emphasizes robust clinical pharmacology characterization, including absorption, distribution, metabolism, and excretion (ADME) profiling. Special attention is given to bioavailability, dose proportionality, and the impact of formulation changes. For peptides developed via the 505(b)(2) pathway, bridging studies and reliance on published literature or reference products may be permitted, provided bioequivalence or clinical comparability is adequately demonstrated.[23]
3.5 Chemistry, Manufacturing, and Controls (CMC)
CMC requirements for peptide therapeutics constitute one of the most technically demanding aspects of FDA review. Sponsors are expected to provide comprehensive documentation covering raw material control, synthetic or recombinant manufacturing processes, purification strategies, in-process controls, and final product specifications. Impurity profiling, including sequence-related impurities, residual solvents, elemental impurities, and degradation products, must be rigorously characterized and controlled. The FDA strongly encourages the application of Quality by Design (QbD) principles in peptide development, including identification of critical quality attributes (CQAs), critical process parameters (CPPs), and establishment of a robust control strategy. Analytical method validation, stability studies under ICH conditions, and comparability assessments following manufacturing changes are essential to ensure product consistency throughout the lifecycle.[24]
3.6 Marketing Authorization Pathways
Peptide therapeutics in the United States may be approved via multiple regulatory pathways depending on their classification and development strategy. Novel peptides are typically approved through the 505(b)(1) NDA pathway, requiring a full complement of nonclinical and clinical data. Modified or reformulated peptides may qualify for the 505(b)(2) pathway, allowing partial reliance on existing data and literature. Unlike biologics, peptide drugs approved under the FD&C Act are not eligible for biosimilar approval under section 351(k) of the PHS Act. Instead, follow-on peptides are regulated as generic drugs under the Abbreviated New Drug Application (ANDA) pathway, provided they meet bioequivalence and pharmaceutical equivalence criteria. This regulatory distinction has significant implications for market competition and intellectual property strategy.[25]
3.7 Post-Marketing Requirements
Post-approval obligations for peptide therapeutics include pharmacovigilance, adverse event reporting, and post-marketing studies as required by the FDA. Manufacturing changes are subject to established comparability and reporting requirements, with the extent of regulatory oversight determined by the potential impact on product quality and clinical performance. Risk Evaluation and Mitigation Strategies (REMS) may be imposed for peptides associated with significant safety concerns, although this is less common compared to high-risk small molecules and biologics. Continuous lifecycle management, supported by real-world evidence and post-marketing surveillance, is increasingly emphasized to ensure long-term safety and efficacy.[26]
4. REGULATORY FRAMEWORK IN INDIA
4.1 Regulatory Authority
In India, peptide therapeutics are regulated by the Central Drugs Standard Control Organization (CDSCO) under the Ministry of Health and Family Welfare. The Drug Controller General of India (DCGI) serves as the national regulatory authority responsible for granting approvals for clinical trials, new drugs, and marketing authorizations. Regulatory oversight is primarily governed by the Drugs and Cosmetics Act, 1940 and the Drugs and Cosmetics Rules, 1945, with recent amendments introducing a more structured framework for new drugs and clinical trials. Peptide therapeutics are generally regulated as “new drugs” under the New Drugs and Clinical Trials (NDCT) Rules, 2019. Unlike the United States, where molecular size and synthetic origin play a defining role in regulatory classification, the Indian regulatory framework adopts a more conservative and process-driven approach. Peptides are frequently evaluated using principles analogous to those applied to biologics, particularly when manufacturing involves recombinant technologies or when product complexity raises concerns regarding immunogenicity and consistency.[27]
4.2 Regulatory Guidelines Relevant to Peptides
India currently lacks peptide-specific regulatory guidance, resulting in reliance on a patchwork of general drug, biologic, and biosimilar guidelines. CDSCO applies relevant ICH guidelines, to which India is a signatory, including ICH Q6B, ICH S6(R1), and ICH Q8–Q11, alongside national documents such as the “Guidelines on Similar Biologics” and various CDSCO circulars and gazette notifications. This absence of explicit peptide-focused guidance introduces interpretative variability and regulatory uncertainty, particularly for chemically synthesized peptides that would otherwise be classified as small molecules in other jurisdictions. Consequently, sponsors are often required to engage in early scientific advice meetings with CDSCO to clarify regulatory expectations, especially for novel or complex peptide products.[28]
4.3 Preclinical Requirements
Preclinical development of peptide therapeutics in India is conducted in accordance with Schedule Y (where applicable) and the NDCT Rules, 2019. Toxicological evaluation typically includes single-dose and repeat-dose toxicity studies, safety pharmacology, local tolerance studies, and, where relevant, reproductive and genotoxicity assessments. For peptides with structural similarity to endogenous compounds, CDSCO may permit a reduced nonclinical package; however, such concessions are granted on a case-by-case basis. Immunogenicity assessment is of particular regulatory concern in India. CDSCO frequently requires immunotoxicity evaluation for peptide products, especially those intended for chronic administration or involving novel sequences. In vivo immunogenicity studies and antibody formation assessments may be mandated, even when comparable data would not be required in more harmonized jurisdictions. This conservative stance reflects a precautionary regulatory philosophy aimed at minimizing long-term safety risks.[29]
4.4 Clinical Trial Requirements
Clinical trials for peptide therapeutics in India require prior approval from CDSCO and registration with the Clinical Trials Registry of India (CTRI). The NDCT Rules, 2019 mandate a phased clinical development approach, although waivers for Phase I or bridging studies may be granted for products approved in certain regulated markets. Local clinical trial requirements represent a key regulatory divergence between India and the United States. Even when extensive global clinical data are available, CDSCO may require India-specific clinical studies to assess ethnic sensitivity, safety, and efficacy. Study design must comply with Good Clinical Practice (GCP) guidelines, and ethics committee approval is mandatory for all trials.[30]
4.5 Chemistry, Manufacturing, and Controls (CMC) Requirements
CMC evaluation constitutes a critical component of CDSCO’s regulatory review for peptide therapeutics. Sponsors are expected to provide detailed information on raw material sourcing, synthesis or expression systems, purification processes, in-process controls, and finished product specifications. Emphasis is placed on impurity characterization, batch-to-batch consistency, and stability under Indian climatic conditions. CDSCO often applies biologics-oriented expectations to peptide CMC dossiers, particularly for recombinant or complex synthetic peptides. Comparability assessments following manufacturing changes are scrutinized rigorously, and post-approval changes may require prior regulatory approval rather than notification. The application of Quality by Design (QbD) principles, although encouraged, is not yet uniformly implemented, leading to variability in regulatory acceptance.[31]
4.6 Marketing Authorization Pathways
Marketing authorization for peptide therapeutics in India is granted through the New Drug Application process under the NDCT Rules, 2019. Unlike the FDA, CDSCO does not provide a distinct regulatory pathway analogous to the 505(b)(2) framework. As a result, reliance on published literature or reference products is limited, and sponsors are often required to submit comprehensive nonclinical and clinical datasets. Follow-on or generic versions of peptide therapeutics are not subject to a clearly defined abbreviated pathway. In practice, such products may be evaluated under frameworks similar to those applied to biosimilars, requiring extensive comparability data. This regulatory ambiguity poses challenges for market entry and may discourage investment in follow-on peptide development.[32]
4.7 Post-Marketing Regulations
Post-marketing surveillance in India is overseen through the Pharmacovigilance Programme of India (PvPI). Marketing authorization holders are required to report adverse drug reactions, submit periodic safety update reports (PSURs), and comply with post-marketing study requirements when mandated. Manufacturing changes, formulation modifications, and site transfers are subject to CDSCO approval, with limited flexibility for post-approval change management. This stringent oversight, while ensuring patient safety, can prolong regulatory timelines and increase operational burden for sponsors. Strengthening risk-based lifecycle management frameworks remains an area of ongoing regulatory evolution in India. [33]
5. COMPARATIVE ANALYSIS: USA VS INDIA
The regulatory oversight of peptide therapeutics in the United States and India reflects fundamentally different philosophical approaches to product classification, risk assessment, and lifecycle management. While both jurisdictions aim to ensure safety, efficacy, and quality, the criteria used to evaluate peptide-based drugs diverge substantially, creating challenges for sponsors pursuing parallel or sequential global development.
Table 2. Comparative Regulatory Analysis of Peptide Therapeutics in the USA and India. [20,22,23,24,26,28,29,30,32]
|
Regulatory Aspect |
United States (FDA) |
India (CDSCO) |
|
Regulatory Authority |
U.S. Food and Drug Administration (FDA), primarily through CDER |
Central Drugs Standard Control Organization (CDSCO) under DCGI |
|
Governing Legislation |
Federal Food, Drug, and Cosmetic (FD&C) Act; Public Health Service (PHS) Act (limited cases) |
Drugs and Cosmetics Act, 1940; NDCT Rules, 2019 |
|
Product Classification Criteria |
Molecule-centric and risk-based; chemically synthesized peptides (<40 amino acids) typically regulated as small molecules |
Process-centric and conservative; peptides frequently regulated under biologics-like frameworks regardless of size |
|
Regulatory Paradigm |
Flexible, science-driven, case-by-case assessment |
Precautionary, process-driven, and relatively prescriptive |
|
Availability of Peptide-Specific Guidance |
No dedicated peptide-specific guidance; evaluated using hybrid small-molecule and biologics frameworks |
No peptide-specific guidance; reliance on general drug, biologic, and biosimilar guidelines |
|
Applicable ICH Guidelines |
ICH Q6A/Q6B, S6(R1), Q8–Q12 (fully adopted and operationalized) |
ICH Q6B, S6(R1), Q8–Q11 (adopted but variably implemented) |
|
Preclinical Toxicology Requirements |
Risk-based; reduced nonclinical package may be accepted for endogenous or low-risk peptides |
Generally conservative; broader toxicology and immunotoxicity studies frequently required |
|
Immunogenicity Assessment |
Risk-based evaluation; reduced testing possible with scientific justification |
Often mandatory in vivo immunogenicity and antibody formation studies |
|
IND / Clinical Trial Authorization |
Investigational New Drug (IND) application |
CDSCO approval + CTRI registration |
|
Clinical Trial Flexibility |
Adaptive designs and global data acceptance permitted |
Limited flexibility; India-specific clinical trials often required |
|
Reliance on Foreign Clinical Data |
Permitted, especially under 505(b)(2) pathway |
Restricted; waivers possible but inconsistently granted |
|
CMC Expectations |
Strong emphasis on QbD, impurity profiling, and lifecycle management |
Emphasis on batch consistency, impurity control, and climatic stability |
|
Post-Approval Change Management |
Risk-based reporting; comparability protocols widely accepted |
Prior approval often required for manufacturing changes |
|
Marketing Authorization Pathways |
NDA (505(b)(1)), NDA (505(b)(2)), ANDA for follow-on peptides |
New Drug Application under NDCT Rules, 2019 |
|
Follow-On / Generic Peptide Pathway |
Clear ANDA pathway based on pharmaceutical equivalence and bioequivalence |
No defined abbreviated pathway; often treated akin to biosimilars |
|
Biosimilar Framework Applicability |
Not applicable for peptides regulated under FD&C Act |
Frequently applied in practice for follow-on peptides |
|
Post-Marketing Surveillance |
FDA Adverse Event Reporting System (FAERS), REMS where applicable |
Pharmacovigilance Programme of India (PvPI), mandatory PSURs |
|
Regulatory Timelines |
Relatively predictable and streamlined |
Longer and less predictable due to discretionary requirements |
|
Overall Regulatory Maturity for Peptides |
High regulatory clarity and flexibility |
Moderate; evolving but fragmented |
6. CASE STUDIES
Case studies provide practical insight into how regulatory frameworks for peptide therapeutics are interpreted and applied in real-world development and approval scenarios. Comparative evaluation of selected peptide products approved in the United States and India highlights the operational consequences of divergent classification logic, clinical requirements, and CMC expectations.
Table 3. Case studies highlighting regulatory differences in peptide therapeutics.[34,37,38]
|
Peptide class |
Example product |
USA regulatory pathway |
India regulatory pathway |
Key regulatory insight |
|
GLP-1 receptor agonists |
Semaglutide |
NDA (505(b)(1)) |
NDA with local data |
India requires additional clinical evidence |
|
GnRH analogs |
Leuprolide |
NDA → ANDA for generics |
Biologic-like review |
Formulation complexity drives scrutiny |
|
Somatostatin analogs |
Octreotide |
NDA with comparability |
Stringent post-approval controls |
Lifecycle rigidity in India |
6.1 GLP-1 Receptor Agonists: Metabolic Peptides
Glucagon-like peptide-1 (GLP-1) receptor agonists represent one of the most successful classes of peptide therapeutics globally. In the United States, products such as exenatide, liraglutide, and semaglutide were approved through NDA pathways under the FD&C Act, with regulatory evaluation emphasizing pharmacokinetics, dose–response characterization, and long-term safety, particularly cardiovascular risk.[34] The FDA permitted reliance on extensive global clinical data, post-marketing commitments, and adaptive trial designs to support approval. In India, the approval of GLP-1 receptor agonists followed a more conservative regulatory trajectory. CDSCO required additional local clinical data and imposed stringent CMC scrutiny, particularly for lipidated peptide analogs with extended half-lives. Stability testing under zone IVb climatic conditions and enhanced impurity characterization were emphasized. These requirements contributed to longer approval timelines compared to the United States, despite the availability of robust international data.[35]
6.2 GnRH Analogs: Endocrine and Oncology Applications
Gonadotropin-releasing hormone (GnRH) analogs, such as leuprolide and goserelin, provide a representative example of long-established peptide therapeutics with extensive clinical histories.[36] In the United States, these products were approved as small-molecule drugs and subsequently supported generic competition through the ANDA pathway. Bioequivalence and pharmaceutical equivalence served as the primary approval criteria for follow-on versions, enabling efficient market entry. In India, GnRH analogs have been subject to regulatory treatment resembling that of biologics, particularly for depot formulations. CDSCO frequently required comparative clinical studies and detailed release kinetics data to address concerns related to long-acting delivery systems. This approach reflects heightened regulatory sensitivity to formulation complexity rather than molecular structure alone.[37]
6.3 Somatostatin Analogs: Long-Acting Peptide Formulations
Somatostatin analogs, including octreotide and lanreotide, illustrate regulatory challenges associated with long-acting peptide formulations. The FDA evaluated these products using a risk-based framework, emphasizing in vitro release testing, pharmacokinetic comparability, and post-marketing surveillance to manage long-term safety risks. Manufacturing changes were often managed through established comparability protocols. In India, somatostatin analogs encountered more restrictive regulatory oversight, with CDSCO requiring additional clinical data and extensive stability studies. Post-approval manufacturing changes were subject to prior approval, limiting operational flexibility. These case studies underscore the impact of differing lifecycle management philosophies on product sustainability.[38]Collectively, these case studies demonstrate that regulatory divergence between the United States and India extends beyond formal guidelines to practical implementation. FDA’s flexible, science-driven approach facilitates innovation and lifecycle optimization, whereas CDSCO’s conservative stance prioritizes risk mitigation, often at the expense of regulatory efficiency. For sponsors, these examples highlight the importance of tailoring development strategies to jurisdiction-specific expectations. Early alignment of CMC and clinical plans with regional regulatory philosophies is essential to minimizing delays and ensuring successful market entry for peptide therapeutics.
7. CHALLENGES AND GAPS
Despite the increasing clinical and commercial prominence of peptide therapeutics, significant regulatory challenges persist due to their ambiguous classification and the absence of peptide-specific regulatory guidance. In both the United States and India, peptides are evaluated under hybrid small-molecule and biologics frameworks that inadequately address their unique structural and immunological characteristics, resulting in interpretational variability and increased evidentiary burden on sponsors. Divergent classification approaches risk-based in the United States and process-centric in India further create inconsistencies in regulatory treatment, complicating global development strategies and follow-on product approval pathways. Variability in immunogenicity assessment standards, lack of clarity regarding the clinical relevance of low-titer antibodies, and differences in CMC lifecycle management particularly in post-approval change flexibility and Quality by Design implementation exacerbate regulatory inefficiencies. Additionally, mandatory local clinical trial requirements in India, despite robust global data, raise concerns regarding scientific redundancy and ethical proportionality. These systemic gaps, compounded by limited specialized regulatory expertise, collectively impede streamlined development, lifecycle optimization, and timely patient access to peptide therapeutics.[39,40]
8. EMERGING TRENDS AND FUTURE PERSPECTIVES
The regulatory framework governing peptide therapeutics is undergoing strategic transformation in response to rapid innovation in peptide engineering, manufacturing science, and clinical deployment. The expansion of advanced modalities including peptide–drug conjugates, stapled and lipidated peptides, and long-acting depot systems has exposed the limitations of legacy classification models, necessitating modernized, science-aligned regulatory guidance capable of addressing hybrid constructs that transcend traditional small-molecule and biologic boundaries. Concurrently, advancements in solid-phase synthesis, continuous manufacturing, and high-resolution analytical platforms are driving a paradigm shift toward enhanced process understanding, real-time release testing, and robust Quality by Design implementation.[41] Global regulatory convergence, particularly through strengthened ICH alignment and collaborative harmonization initiatives, is anticipated to reduce duplicative requirements and enhance predictability, while formal peptide-specific guidance would further streamline development pathways. The evolution of clear, risk-based frameworks for follow-on and generic peptides will be critical to balancing innovation with market competition and access. Additionally, the integration of real-world evidence, advanced pharmacovigilance systems, and lifecycle-oriented regulatory oversight signals a transition toward adaptive, patient-centric regulation, wherein benefit–risk assessment becomes a dynamic, data-driven continuum rather than a static pre-approval determination.[42]
CONCLUSION
Peptide therapeutics represent a rapidly expanding and scientifically sophisticated class of medicinal products with the potential to address significant unmet clinical needs across a wide range of therapeutic areas. Their intermediate molecular complexity, positioned between small molecules and biologics, confers both pharmacological advantages and regulatory challenges that are not adequately addressed by existing drug approval frameworks. This review highlights substantial divergence between the regulatory approaches adopted by the United States Food and Drug Administration and India’s Central Drugs Standard Control Organization. While the FDA employs a flexible, risk-based, and molecule-centric paradigm that facilitates innovation and efficient lifecycle management, CDSCO adopts a more conservative, process-driven approach that often subjects peptide therapeutics to biologics-like regulatory scrutiny. These differences manifest across multiple dimensions, including product classification, immunogenicity assessment, clinical trial requirements, CMC evaluation, and post-approval change management. Although harmonization efforts through ICH guidelines have established a partial common foundation, their application to peptide therapeutics remains inconsistent and fragmented. The absence of peptide-specific regulatory guidance in both jurisdictions contributes to interpretative variability, regulatory uncertainty, and development inefficiencies, particularly for complex peptides, long-acting formulations, and follow-on products. Addressing these gaps will require targeted regulatory reform, enhanced regulatory science capacity, and greater international collaboration. Future regulatory frameworks should prioritize the development of dedicated, science-based guidance for peptide therapeutics that reflects their unique physicochemical and biological attributes. Greater reliance on Quality by Design principles, risk-based immunogenicity assessment, acceptance of global clinical data, and streamlined post-approval change management will be critical to supporting innovation while maintaining rigorous safety standards. Achieving regulatory convergence in these areas will not only reduce development timelines and costs but also accelerate patient access to safe and effective peptide-based therapies worldwide.
REFERENCES



Praveen R. B., Dr. Vijaya Raghavan, Iswareyaa R. A, A Comprehensive Review of Regulatory Requirements and Approval Pathways for Peptide Therapeutics in the USA and India Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 2740-2755. https://doi.org/10.5281/zenodo.18672906
 10.5281/zenodo.18672906
10.5281/zenodo.18672906