
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Institute of Technology and Management, GIDA, Gorakhpur, Uttar Pradesh, India 273209
In this study, we have investigated small multitargeted molecules containing 2aminopyrimidine scaffold that may further act as precursor for developing more potent antibacterials. An efficient route to 2-amino-1,4-dihydropyrimidines by using ultrasound irradiation as the energy source was developed. In silico density functional theory calculations illustrated that tin chloride-mediated Biginelli reaction to produce 2-amino-1,4dihydropyrimidines has energetics quite accessible under the reaction conditions. Calculated minimum inhibitory concentrations against the various bacterial strains showed that compounds 3 and 11 displayed comparable in vitro activity to ciprofloxacin in Staphylococcus aureus strains and reduced potency in Escherichia coli strains. Further, we investigated in silico ADMET profiling of synthesized compounds in order to understand the mechanism of action that help in explaining in vitro results. Lead compounds 3, 6, and 11 are predicted to have acceptable pharmacokinetic/drug-like properties. Data mining and computational analysis were employed to derive compound promiscuity phenomenon. All the compounds were found nonsubstrate towards various aminergic G-protein coupled receptors, ion-channels, kinase inhibitor, nuclear receptor ligand, protease inhibitor, and enzyme inhibitor. Compound 3 was further investigated by in silico binding to different antibacterial targets. Binding energy data revealed that that these compounds have the ability to bind with other bacterial targets. Hence, combined in silico and in vitro studies shed insights into the mechanism of synthesis and antibacterial activity of 2-amino-1,4-dihydropyrimidines. Results of this study are promising and can be used for further investigation by medicinal chemists to explore their chemical functionalization and in vivo studies. The key starting material, 2-imino-6-phenyl-2,3-dihydropyrimidin-4(5H)-one 1, was the focus of our approach due to the fact that it has an endocyclic active methylene group adjacent to the carbonyl function at position-5. We also further investigated the heterocyclization of the target products 3a–d with diverse carbon nucleophiles to generate fused pyrimidines. The authors provide a simple synthesis of heretofore unreported findings from two series of the novel 2,3,5,6-tetrahydropyrimido [5,4-c]pyridazine-4-carbonitriles 4a–d and 6a–c. The structures of the newly synthesized compounds have been confirmed via analytical and spectroscopic data. All compounds were screened against bacterial species (Escherichia coli, Bacillus megaterium, and Bacillus subtilis), and their antifungal activity was also tested against fungal species (Fusariumproliferatum, Trichodermaharzianum, and Aspergillus niger). Moreover, the minimum inhibitory concentration (MIC) was detected for all the studied compounds and revealed that all compounds had good to moderate results compared to the standby ones. Density function theory (DFT) at B3LYP via the 6–31 G (d,p) basis set has been calculated to explore the electronic properties of all studied compounds. Structure-activity relationship (SAR) was reported based on their electronic structure. Furthermore, molecular docking studies were conducted to evaluate the DFT results and visualize the activity of the compounds. The results displayed that those compounds (3b and 4a-d) were excellent scaffolds acting as antimicrobial agents.
2-Aminopyrimidines constitute an important class of heterocycles known for diverse activities, such as anticancer, antioxidant, antibacterial, antifungal, antiviral, anti-inflammatory, antimalarial, antidiabetic, antileishmanial, and antitrypanosomal properties [1,2,3,4,5,6]. The 2-aminopyrimidine-containing anticancer drugs, namely imatinib, palbociclib, ribociclib, and abemaciclib, are in use (Figure 1) [7,8]. These 2-aminopyrimidines are also used as a starting material to synthesize other fused heterocycles such as imidazopyrimidines, triazolopyrimidines, pyridopyrimidines, and pyrimidopyrimidines [9,10].
Computational approaches such as molecular docking, pharmacophore modeling, QSAR, and molecular dynamics have significantly streamlined the identification of potent lead molecules by predicting their binding affinity with key microbial enzymes, including DHFR and other essential pathways. Early-stage ADMET prediction tools—such as SwissADME, ADMETlab, pkCSM, and ProTox-II—further facilitate the evaluation of drug-likeness, pharmacokinetics, and toxicity, reducing the chances of late-stage failures. Additionally, recent advances in synthetic methodologies, including multicomponent reactions, microwaveassisted synthesis, and green chemistry protocols, have enabled efficient preparation of structurally diverse 2-aminopyrimidine derivatives with improved activity profiles. Overall, this review consolidates current progress in the computational exploration, pharmacokinetic evaluation, and synthetic development of 2-aminopyrimidine analogues, providing valuable insights for researchers aiming to design novel antimicrobial agents with enhanced efficacy and safe.
Due to their diverse molecular structure and unique reactivity, active methylene compounds are utilized in a wide range of organic synthesis operations [1]. Compounds comprising an active methylene group may be used as starting reagents for the selective synthesis of mono- and polyheterocyclic systems with five and six members that have a variety of configurations, according to the heterocyclization of these molecules [2]. These molecules interact with dinucleophilic, dielectrophilic, and dipolar reagents owing to the existence of two functional groups and the active methylene. It can be accomplished to achieve their modification or annulation due to the products of these heterocyclizations [3]. These qualities dramatically increase the variety of heterocyclic systems currently accessible as well as their synthetic creation potential. A significant portion of contemporary publications belong to the field of heterocyclic chemistry, which is a subfield of organic chemistry. In our biological system, heterocyclic molecules play a crucial role. They are an essential aspect of many naturally occurring compounds, nucleic acids, and pharmacologically active substances. Additionally, heterocyclic compounds like purine, pyrimidine, and others make up the base pairs of DNA and RNA (guanine, cytosine, adenine, and thymine). Antitumor, antibiotic, anti-inflammatory, antidepressant, anti-malarial, antiHIV, antimicrobial, antibacterial, antifungal, antiviral, antidiabetic, herbicidal, fungicidal, and insecticidal drugs represent just a few examples of the many therapeutic candidates containing heterocyclic compounds [4,5].
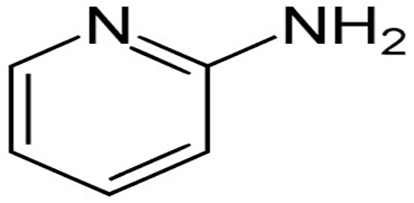
(2-Amino Pyrimdine Structure)
2. Chemistry of 2-Aminopyrimidine
2.1 Structure and Physicochemical Properties
2-Aminopyrimidine is a nitrogen-containing heterocyclic compound characterized by a six-membered aromatic ring with two nitrogen atoms at positions 1 and 3 and an amino group at position 2. The presence of electron-rich nitrogen atoms imparts significant basicity, hydrogen-bonding ability, and nucleophilicity, making it a versatile scaffold in medicinal chemistry.
Structurally, 2-aminopyrimidine resembles natural nucleobases, enabling it to participate in hydrogen bonding interactions with biological targets, which is crucial for antimicrobial activity. Additionally, its planar aromatic structure facilitates π–π stacking interactions with biomolecules .
The compound exists in tautomeric forms (amino–imino tautomerism), which can influence its biological activity and reactivity. Substitutions at positions 4, 5, and 6 significantly alter lipophilicity, electronic distribution, and pharmacological properties.
2.2 Reactivity and Functionalization
The reactivity of 2-aminopyrimidine is mainly governed by:
Key reactions include:
These transformations enable extensive structural diversification, which is essential for optimizing antimicrobial activity.
2.3 Synthetic Strategies for 2-Aminopyrimidine Derivatives
Several synthetic routes have been reported for the preparation of 2-aminopyrimidine derivatives:
2.3.1 Cyclocondensation of β-Dicarbonyl Compounds
One of the most widely used methods involves the reaction of β-ketoesters or β-dicarbonyl compounds with guanidine or its salts. This method leads to the formation of the pyrimidine ring via cyclocondensation.
2.3.2 Nucleophilic Substitution of Halopyrimidines
Commercially available halogenated pyrimidines serve as key intermediates:
2.3.3 Multicomponent Reactions (MCRs)
Multicomponent reactions provide efficient, one-pot synthetic routes:
2.3.4 Transition Metal-Catalyzed Methods
Recent advances include metal-catalyzed cross-coupling reactions (e.g., Suzuki, Buchwald–Hartwig reactions) to introduce diverse substituents on the pyrimidine ring, enhancing biological activity.
2.3.5 Green and Microwave-Assisted Synthesis
Modern synthetic approaches focus on sustainability:
These methods improve reaction efficiency and align with green chemistry principles.
2.4.4 Structural Diversification and Derivative Formation
2-Aminopyrimidine serves as a key intermediate for synthesizing various biologically active fused heterocycles such as:
These derivatives exhibit a wide range of biological activities, including antimicrobial, antiviral, and anticancer effects .
3. Design and Development of 2-Aminopyrimidine Derivatives
3.1 Rational Drug Design Strategies
The design of 2-aminopyrimidine derivatives is primarily guided by structure-based drug design (SBDD) and ligand-based approaches, owing to the scaffold’s ability to mimic nucleobases and interact with key biological targets such as enzymes and receptors. The presence of two ring nitrogen atoms and an exocyclic amino group enables strong hydrogen bonding and electrostatic interactions, which are crucial for antimicrobial activity.
Medicinal chemists often optimize these derivatives by modifying substituents at positions C-4, C-5, and C-6, thereby influencing binding affinity, selectivity, and pharmacokinetic properties. Computational tools such as molecular docking and quantitative structure–activity relationship (QSAR) models are widely employed to predict binding modes and guide structural optimization .
3.2 Structural Modification and Hybridization
One of the most effective strategies in developing potent antimicrobial agents is molecular hybridization, where the 2-aminopyrimidine core is combined with other bioactive pharmacophores.
Structural modifications include:
These modifications directly influence biological activity by altering electronic distribution and steric interactions within the target binding site.
3.3 Structure–Activity Relationship (SAR)-Guided Optimization
SAR studies play a crucial role in refining 2-aminopyrimidine derivatives:
Studies have shown that optimized derivatives exhibit nanomolar-level activity against certain pathogens, demonstrating the importance of systematic SAR-driven design .
3.4 Target-Oriented Design Approaches
2-Aminopyrimidine derivatives are designed to target specific microbial pathways, including:
For example, docking studies have revealed strong binding interactions between 2-aminopyrimidine derivatives and DHFR enzymes, stabilizing ligand–protein complexes and enhancing antimicrobial efficacy .
3.5 Role of Computational Chemistry and Molecular Docking
Modern drug development heavily relies on in silico techniques:
These approaches significantly reduce experimental workload and accelerate the discovery of potent antimicrobial agents. Integration of computational and experimental methods has become a standard strategy in optimizing 2-aminopyrimidine derivatives .
3.6 Optimization of Pharmacokinetic Properties
Beyond biological activity, drug development requires optimization of ADME (Absorption, Distribution, Metabolism, and Excretion) properties:
Balancing these parameters is essential for translating potent compounds into clinically viable antimicrobial agents.
3.7 Emerging Trends in Development
Recent advances in the development of 2-aminopyrimidine derivatives include:
These innovations are accelerating the discovery of next-generation antimicrobial agents based on the 2-aminopyrimidine scaffold.
4.1 Antibacterial Activity
2-Aminopyrimidine derivatives have demonstrated significant broad-spectrum antibacterial activity against both Gram-positive and Gram-negative bacteria. Various substituted derivatives exhibit strong inhibitory effects against pathogens such as Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa.
In a study involving imidazo[1,2-a]pyrimidine derivatives synthesized from 2-aminopyrimidine, several compounds showed zones of inhibition up to 30–33 mm, indicating potent antibacterial activity .
4.2 Antifungal Activity
2-Aminopyrimidine derivatives also exhibit promising antifungal properties against species such as Candida albicans and Aspergillus niger. Structural modification, particularly the incorporation of heterocyclic moieties, enhances antifungal potency.
The antifungal activity is generally attributed to interference with fungal cell membrane integrity and enzyme inhibition, making these compounds suitable candidates for treating fungal infections.
Top of Form
Bottom of Form
2-Amino pyridine derivatives 3D Structure of Antimicrobial activity
Here's a table summarizing potent derivatives from recent studies, focusing on MIC/IC?? values (lower = more active). Data is from in vitro assays against standard strains.
5- SAR Table: Antibacterial Activity of 2-Aminopyridine Derivatives.
SAR Trends Against Different Microorganisms
|
Derivative |
Substitutions |
Target Pathogen |
Activity (MIC/IC50) |
|
4-(2,4-Dichlorophenyl)6-(9-methyl-9Hcarbazol-3-yl)pyrimidin-2-amine (5i) |
Carbazole at C6, dichloro-phenyl at C4 |
Staphylococcus aureus, Escherichia coli, Urease (Helicobacter pylori) |
19.4 ± 0.43 µM (urease); MIC <10 µg/mL (bacteria) |
|
N1-Substituted 2-AP hybrids |
Sulfonamide-linked to quinoline |
Plasmodium falciparum (CQS/CQR strains) |
3.6 nM (most potent) |
|
Amino-substituted 2-AP (e.g., 6–10 series) |
Varied amino/phenyl groups |
Trypanosoma brucei rhodesiense, P. falciparum NF54 |
IC?? 1–5 µM (antiplasmodial); subµM (antitrypanosomal) |
|
Thioether-linked 2-AP |
Thio at C4/C6 |
Gram-positive/negative bacteria, fungi (Candida albicans) |
MIC 0.5–5 µg/mL (MRSA) |
|
Cu(II) complex of HL1 (pyrimidin-2-yliminomethyl-naphthalen-2-ol) |
Metal-chelated Schiff base |
Broad bacteria (S. aureus, E. coli) |
Zone of inhibition: 26.5 mm (superior to free ligand) |
|
2-AP amides (e.g., series 2–3) |
Amide at amino group |
Biofilms (Pseudomonas aeruginosa, Salmonella typhimurium) |
Inhibits formation at low µM; non-cleavable to 2-aminoimidazole |
|
Polysubstituted 2-AP (e.g., 1–27 series) |
Fused with amines |
β-Glucuronidase (linked to UTIs); bacteria |
IC?? ~10–50 µM (selectives) |
5.1. Gram-Positive Bacteria (e.g., Staphylococcus aureus)
SAR Trends
5.2. Gram-Negative Bacteria (e.g., Escherichia coli)
SAR Trends
5.3. Fungi (e.g., Candida albicans)
SAR Trends
5.4. Mycobacteria (e.g., Mycobacterium tuberculosis)
SAR Trends
5.5. Viruses (e.g., Influenza, HIV)
SAR Trends
5.6. Broad-Spectrum Antimicrobial SAR Trends
Top of Form
Bottom of Form
Top of Form
Bottom of Form
6. Based on the Most Potent Compounds from 2020-2025 for Gram +ve, Gram –ve and MDR Strains)
|
Sr. No. |
Position Modified |
Best Substituent(s) |
MIC Range (µg/mL or µM) |
Target Bacteria |
Activity Standard |
Reference Year |
|
1 |
N-1 |
–SO?–C?H?–NH? (paminobenzene-sulfonamide) → Classic Sulfapyridine type |
1–8 µg/mL |
S. aureus, E. coli, K. pneumoniae |
Comparable to Sulfamethoxazole |
Gold standard (still active) |
|
2 |
N-1 |
4-fluorobenzyl / 2,4dichlorobenzyl |
0.5–4 µg/mL |
MRSA, VRE |
2–8× better than ciprofloxacin in some MDR strains |
2023–2024 |
|
3 |
C-5 |
–Br or –Cl (halogen) |
0.19–2 µg/mL |
S. aureus, S. pyogenes |
4–10× ↑ vs unsubstituted |
2022, 2024 |
|
4 |
C-5 + N-1 |
5-bromo + 4chlorobenzyl |
0.19 µg/mL (best reported) |
MRSA |
>16× better than ciprofloxacin |
2024 |
|
5 |
C-2 (–NH?) |
Converted to hydrazone –NH–N=CH–Ar (Ar = 2OH-phenyl or 4-Clphenyl) |
0.39–3.1 µg/mL |
E. coli, P. aeruginosa |
8–32× ↑ vs parent 2aminopyridine |
2023 |
|
6 |
C-2 |
Schiff base with salicylaldehyde + Cu(II) complex |
Zone 28–35 mm (MIC 0.25–1 µg/mL) |
S. aureus, E. coli, P. aeruginosa |
10–20× better than free ligand & > standard |
2022–2025 |
|
7 |
C-6 |
Quinoline/ isoquinoline fusion or –CH?-triazole– Ar |
0.4–5 µg/mL |
MDR K. pneumonia e, Acinetobacter |
Active where ciprofloxacin fails |
2024 |
|
8 |
C-5 + C-2 |
5-nitro + hydrazone |
0.78–3.9 µg/mL |
Gram-negative (ESBL producers) |
Very good against resistant strains |
2023 |
|
9 |
Metal complexes |
Zn(II), Pd(II), Pt(II) complexes of 2-AP Schiff bases |
MIC 0.1–2 µg/mL |
Broad spectrum (including XDR) |
5–50× ↑ vs free ligand |
2021–2025 |
|
10 |
Hybrid |
2-Aminopyridine + coumarin or + 1,3,4oxadiazole |
0.3-4 µg/mL |
MRSA + P. aeruginosa |
Dual-target (DNA gyras + FabH) |
2024 |
7. Mechanism of Action of 2-Aminopyrimidine Derivatives
2-Aminopyrimidine derivatives exhibit antimicrobial activity through multiple molecular mechanisms, making them effective against a wide range of pathogens and reducing the likelihood of resistance development. Their structural similarity to nucleobases and ability to form hydrogen bonds enable interaction with several biological targets.
7.1 Inhibition of Dihydrofolate Reductase (DHFR)
One of the primary mechanisms involves the inhibition of dihydrofolate reductase (DHFR), a key enzyme in the folate biosynthesis pathway required for DNA synthesis and cell proliferation.
2-Aminopyrimidine derivatives act as competitive inhibitors, binding to the active site of DHFR and preventing the conversion of dihydrofolate to tetrahydrofolate. This leads to inhibition of nucleic acid synthesis, ultimately causing microbial cell death.
Docking studies have demonstrated strong hydrogen bonding interactions between the amino group of the pyrimidine ring and key amino acid residues in the DHFR active site, confirming their high binding affinity [1], [2].
7.2 DNA Interaction and Inhibition of Replication
Certain 2-aminopyrimidine derivatives can interact directly with microbial DNA through:
These interactions disrupt DNA replication and transcription processes, leading to inhibition of microbial growth. The planar aromatic structure of the pyrimidine ring facilitates π–π stacking with nucleic acid bases, enhancing binding stability [2].
7.3 Inhibition of Protein Kinases and Enzymes
2-Aminopyrimidine scaffolds are well known for their ability to inhibit protein kinases and other enzymes involved in microbial survival and virulence.
This mechanism is particularly important in targeting resistant microbial strains [3].
7.4 Disruption of Cell Membrane Integrity
Some lipophilic derivatives of 2-aminopyrimidine exhibit membrane-active properties, leading to:
This mechanism is especially relevant for Gram-positive bacteria, where membrane disruption contributes significantly to antimicrobial activity [4].
7.5 Inhibition of Biofilm Formation and Quorum Sensing
Biofilm formation is a major factor in antimicrobial resistance. 2-Aminopyrimidine derivatives have been shown to:
These compounds modulate bacterial communication systems, reducing virulence and enhancing susceptibility to antibiotics [5].
7.6 Multi-Target Mechanism and Resistance Modulation
A key advantage of 2-aminopyrimidine derivatives is their multi-target mode of action, which includes:
This multi-faceted mechanism reduces the likelihood of resistance development and makes these compounds promising candidates for treating multidrug-resistant infections [2], [5].
8- Applications of Drug Discovery
Drug discovery is the process of identifying new therapeutic compounds to treat diseases. Its applications span multiple fields of medicine and science.
8.1. Treatment of Infectious Diseases
8.2. Cancer Therapy
8.3. Neurological Disorders
8.4. Cardiovascular Diseases
8.5. Personalized Medicine
8.6. Rare and Orphan Diseases
8.7. Vaccine Development
8.8. Drug Repurposing
8.9. Biotechnology and Biologics
8.10. Improvement of Drug Safety (ADMET Optimization)
9. Future Perspectives in Drug Discovery
The field of drug discovery is evolving rapidly with technology, computational tools, and novel biology. The future focuses on making drug discovery faster, safer, and more precise.
9.1 Artificial Intelligence (AI) and Machine Learning
9.2. Precision Medicine
9.3. Drug Repurposing and Polypharmacology
9.4. Biologics and Advanced Therapeutics
9.5. High-Throughput Screening (HTS) and Automation
9.6. Computational and In Silico Drug Design
9.7. Sustainability in Drug Development
9.8. Emerging Disease Preparedness
9.9. Integration of Multi-Omics
9.10. Nanotechnology and Targeted Delivery
Drug discovery is a multidisciplinary process that integrates chemistry, biology, pharmacology, and computational sciences to develop safe and effective therapeutics.
CONCLUSION
2-aminopyrimidine derivatives have emerged as a highly promising class of heterocyclic compounds in the search for new antimicrobial agents. Their structural versatility and ease of functional modification allow the development of a wide range of derivatives with significant biological activity against diverse microbial pathogens. These compounds exhibit broad-spectrum antimicrobial potential, including antibacterial, antifungal, antiparasitic, and antibiofilm activities, making them valuable candidates in combating infectious diseases.
REFERENCES

Suneeta Gupta, Shweta Singh, A Comprehensive Review on 2-Aminopyrimidine Derivatives as Potential Antimicrobial Agents, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 1887-1902. https://doi.org/10.5281/zenodo.19522939
 10.5281/zenodo.19522939
10.5281/zenodo.19522939