
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmacy, Faculty of Engineering and Technology, MJP Rohilkhand University, Bareilly, Uttar Pradesh, India 243006
Solubility refers to the ability of a solid to dissolve in a liquid and form a consistent solution. Ensuring that there is the proper dosage of medicine in the blood is vital for its effectiveness or desired pharmacological response. A significant challenge in developing new medications is that many of them have poor solubility in water. This is particularly applicable to in BCS Class II and IV. The slow dissolution of these drugs leads to inadequate absorption in the body when they are taken orally. In this review, the necessity of improving solubility is highlighted, along with an explanation of how the solubilization process operates and the main physical and chemical elements that impact it. This review primarily aims to explore methods that enhance the absorption and bioavailability of drugs that are poorly soluble in the body. Various conventional and advanced solubility methods for enhancing solubility are meticulously examined. This includes particle size reduction, pH modification, cosolvency, hydrotropy, solid dispersion, complexation, surfactant-based systems, and nanotechnology-driven approaches. The objective of this review is to discuss a range of approaches to boost the dissolution of substances, ultimately aiming to enhance the performance of drugs.
Enhancing the usability of poorly dissolving drugs poses a common hurdle during the testing phase of new chemical and product creation [1]. Several strategies can facilitate better dissolution and increased effectiveness in the body for drugs that have low solubility in water. Oral medications are completely absorbed only after they dissolve in gastric fluids, ensuring they are readily available for the body to utilize. The bioavailability of a drug is influenced by several factors, primarily its ability to dissolve in water and its ease of permeability through lipophillic membranes [2].The methods generally used for solubilization of drug includes micronization, chemical modification, pH adjustment, solid dispersion, complexation, co-solvency, hydrotropy etc.
In order for drug molecules to access their sites of action in the body, like the bloodstream, they must first be dissolved to pass through cell membranes. A drug needs to be dissolved in a water-based solution in order to be absorbed by the body.[3,4] The absorption of a drug in the body is affected by its ability to dissolve and move through membranes, which can be altered or improved with particular techniques [5]. In the Biopharmaceutical Classification System (BCS), compounds that have poor solubility are categorized as class II. When subjected to laboratory testing, they encounter numerous obstacles, including limited delivery methods and complex dissolution tests that frequently do not align with actual absorption in the body. Recently, more than 40% of newly developed pharmaceuticals are poorly soluble in water. Medications that are poorly soluble in water tend to be absorbed at a slower rate, resulting in unpredictable effects and potential damage to the stomach lining [6].
One of the major obstacles in creating new medications, particularly tablets, is ensuring that drugs dissolve properly and are effectively absorbed by the body after oral consumption. The characteristics of these substances in vivo and in vitro laboratory experiments, combined with the difficulties in achieving consistent results across these environments, often complicate the formulation of many newly developed compounds due to their solubility issues [7,8]
Although pharmaceutical companies have developed methods to manage poorly soluble drugs, those with aqueous solubility of less than 0.1 mg/ml present some unique challenges. This review begins by discussing conventional techniques used to facilitate drug dissolution. These techniques involve adjusting the pH, employing different co- solvency, and reducing the size of particles. Micro-emulsions and self-emulsifying systems represent innovative approaches. These medications represent excellent alternatives for innovative approaches to their dissolution, produced by companies dedicated to providing pharmaceuticals. Conventional techniques for enhancing drug solubility involve particle size reduction, pH, adjustment and incorporating agents such as surfactants and co-solvents, while micro emulsion and self-emulsifying systems are novel approaches.
There are various methods to enhance the solubility of drugs that have poor water solubility. The selection of methods depends on particular elements, such as the drug's properties, the types of additives required, and the necessary medicine form. This review examines various traditional and innovative techniques aimed at enhancing the absorption of poorly soluble drugs when administered orally.
2. SOLUBILITY
Solubility indicates the extent to which a solute can dissolve uniformly in a solvent. The concentration of the substance is frequently assessed by the quantity of it contained within a certain volume of the liquid. A drug's solubility can be influenced by various factors, including the liquid solvent (like ACN, MeOH, or water), the temperature (measured in degrees Celsius or Kelvin), and the pressure within the system [9]. The saturation concentration refers to the maximum quantity of a substance that can dissolve within a system. After reaching this level, increasing the amount of the substance will not enhance its solubility[10].The state of solubility equilibrium is established when both processes take place simultaneously at the same speed. At times, a solution may contain more solute than it typically would, resulting in a state called supersaturation. Under particular circumstances, this leads to the formation of an unstable solution[11]. In table 1, the United States Pharmacopeia (USP) and the British Pharmacopeia (BP) provide a basic categorization of the solubility of compounds of various substances.
Table 1. USP and BP solubility criteria
|
Description |
Part of solvent required per part of solute |
|
Very Soluble |
Less than 1 |
|
Freely Soluble |
From 1 to 10 |
|
Soluble |
From 10 to 30 |
|
Sparingly Soluble |
From 30 to 100 |
|
Slightly soluble |
From 100 to 1000 |
|
Very slightly soluble |
From 1000 to 10,000 |
|
Practically insoluble |
10,000 and over |
3. SOLUBILITY ISSUES IN DRUG DEVELOPMENT
3.1 High Lipophilicity (High logP / Low Polarity)
A major reason behind poor solubility in water is the lipophilic nature of the compound. Substances that possess a high partition coefficient (logP > 3) exhibit poor solubility in water due to their extensive hydrophobic surfaces, which prevent them from forming hydrogen bonds.The lipophilicity of compounds facilitates their movement through membranes and binding to targets, yet it reduces their solubility by requiring less energy for dissolution in water [12]. Modern drug design frequently develops compounds that are poorly soluble in water while they can effectively address diseases, they pose challenges when it comes to liquid formulations.while they can effectively address diseases, they pose challenges when it comes to liquid formulations.This indicates that particular approaches are necessary to ensure their usability.
3.2 Strong Crystal Lattice Energy and Packing (Polymorphism)
The arrangement of solid materials influences their solubility. Compounds that exhibit solid arrangements with densely packed layers, strong inter-molecular bonds, or smooth surfaces are generally associated with a high level of energy that maintains their cohesion.This poses a challenge to their disintegration[13]. Polymorphism complicates the understanding of solubility, as various crystal forms, hydrated states, and solutions can demonstrate significantly varying solubility levels. For instance, ritonavir unexpectedly transformed into a new version, leading to the product's failure and its subsequent removal from the market until it could be reformulated[14]. Failing to regulate and document the solid form of a substance may result in variations in its solubility and overall efficacy across different batches.
3.3 Lack of Ionizable Groups at Physiological pH
Ionization plays a crucial role in how effectively substances dissolve in water, as charged particles interact more readily with water compared to neutral particles. Medications lacking ionizable groups remain neutral across various pH levels in the body, which means their solubility is solely dependent on their affinity for water[15]. The solubility of weak acids and bases can be assessed through their fundamental solubility (S?) and a parameter known as pKa. Nonetheless, if a substance does not transform into a different form in the acidic environment of our stomach, it tends to have low solubility. This suggests that we need to adopt more advanced approaches to improve its dissolution.
3.4 Large Molecular Size and Low Conformational Flexibility
The dimensions and configuration of molecules play a crucial role. In most cases, larger molecules are characterized by an increased number of components that are incompatible with water and exhibit lower polarity, rendering them difficult to dissolve in water[16]. Rigid, flat molecules typically interlock more efficiently in crystalline structures, resulting in lower solubility compared to their more bendable counterparts[17]. The classification of biopharmaceuticals reflects this trend, with a significant number of newly developed large molecules being placed in BCS Class II or IV.
3.5 Particle Size and Morphology
While the solubility of a solid remains constant, the rate at which it dissolves is influenced by the particle size and the extent of their surface area. The Noyes–Whitney equation elucidates this relationship[18]. Reducing the size of materials through micronization and nanonization enhances their surface area. This facilitates a better dissolution rate and more rapid absorption[19]. Molecule morphology (nebulous vs. crystalline, permeable vs. compact) too impacts disintegration. Nanosizing can drastically move forward disintegration but requires stabilizers to avoid accumulation and Ostwald ripening.
3.6 Solid-State Heterogeneity and Impurities
Variations in the solid state, including distinct forms, amorphous components, residual solvents, and trace impurities, can affect a substance's solubility and lead to inconsistent results [20]. Generally, amorphous solids have a higher dissolution rate compared to crystalline solids, yet they lack stability and may revert to a crystalline state, leading to a decrease in their solubility over time. It is essential to conduct thorough testing with powder X-ray diffraction (PXRD), differential scanning calorimetry (DSC), and microscopy to ensure the substance dissolves consistently.
4. IMPORTANCE OF SOLUBILITY IN ORAL DRUG ABSORPTION
Administering medication orally is the most prevalent and simplest method of drug delivery.There are several advantages to this technique, such as higher compliance among patients, lower manufacturing expenses, reduced infection risks, and a quicker process for producing the medication [21]. Currently, more than 80% of medications are administered orally.The absorption, distribution, utilization, and overall characteristics of a drug after it is ingested are highly influenced by its ability to dissolve. Medications that have poor solubility typically require larger doses or more frequent administration to achieve the desired blood concentration when taken orally. Enhancing the dissolution properties of these drugs is crucial for their development [22].
Improving a drug’s dissolution usually relies on specific aspects of the drug itself, the ingredients, the administration method, and its biological effectiveness in treatment. The effectiveness of a drug and its availability can differ due to the fact that some drugs do not dissolve easily in water. This is particularly relevant for drugs that do not interact well with the fluids found in our gastrointestinal tract. The classification of drug solubility is structured into four main groups, referred to as the Biopharmaceutics Classification System (BCS I-IV).This system illustrates how efficiently a substance can dissolve and move across membranes [23,24]. Improved solubility for BCS class II medications can enhance the drug's bioavailability and accelerate its dissolution in the stomach and intestines.The main challenge with BCS Class II drugs is how effectively they dissolve in the gastric fluid , rather than the ease with which they are absorbed into the body. Furthermore, enhanced dissolution of a drug leads to increased availability for the body’s absorption [25,26].
The development of drugs that do not dissolve effectively often involves challenges like significant research expenditures, prolonged product development times, issues of effectiveness, toxicity concerns, and the degree to which they interact with other ingredients[27].
5. PROCESS OF SOLUBILIZATION
Solubilization involves the disruption of bonds between ions or molecules within the dissolved substance[28]. As the solvent molecules separate, they create space for the solute to fit in.This creates a dynamic interaction between the molecules of the solvent and those of the solute. Solubilization processes are described in below Figure 1.
The process of solubilization involves the breaking of bonds in the solvent, which leads to the emergence of gaps reminiscent of those depicted in Step 1. As described in Step 2, the solubilization process involves external energy breaking the bonds between the molecules of the solid solute. In Step 3, the presence of external energy causes a solid molecule to blend into the liquid.
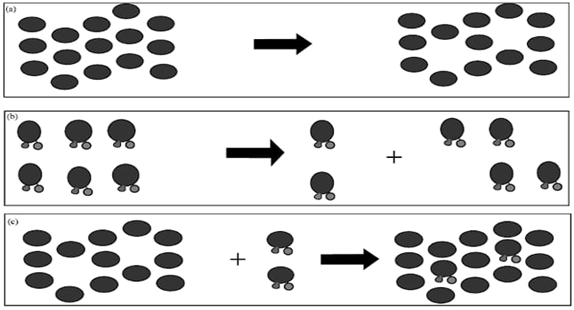
Figure 1. Process of Solubilization [29]
6. FACTORS AFFECTING SOLUBILIZATION
The solubility of a compound is influenced by its physical state (solid, liquid, or gas), the composition of the solvent, the temperature, and the prevailing pressure conditions [30].
Particle Size
The dimension of solid particles plays a crucial role in determining the solubility of a drug.When the size of particles is decreased, the ability of a substance to dissolve increases.As particles decrease in size, their ability to dissolve improves since they possess a larger surface area compared to their overall volume. Larger surface areas on particles result in enhanced interactions with the solvent they are immersed in. The formula below illustrates the impact of particle size on the solubility of a substance.
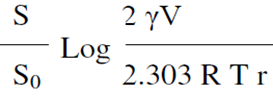
Where S0 is the solubility of infinitely large particles, is the solubility of fine particles, S is solubility of fine particles, V is molar volume, γ is the surface tension of the solid, R is the is the gas constant and T is the temperature.
Temperature
Temperature plays a significant role in determining the solubility of a substance. As the temperature of a mixture increases, the heat energy allows for a quicker interaction between the solute and solvent, promoting their mixing. When the temperature of a solution decreases, it releases energy, making it more difficult for the substances to blend. This indicates that the solution has a lower capacity to dissolve substances. Many solid materials follow this pattern, but there are exceptions among them. Certain solid materials are more soluble as the temperature decreases. For every gas, an increase in the solution's temperature leads to a reduction in its solubility [31].
Molecular Size
The dimensions of a molecule play a crucial role in determining the solubility of a substance. Generally, as molecules increase in size and weight, their solubility tends to decrease. This is due to the fact that larger molecules encounter greater difficulty in being enveloped by solvent molecules. In organic compounds, when carbon extends its branches, it reduces the size of the molecule, enhancing its solubility [32].
Nature of the Solute and Solvent
The quantity of solute is determined by the specific solute and solvent involved. At room temperature, you can dissolve only 1 gram of lead (II) chloride or as much as 200 grams of zinc chloride in 100 grams of water.
Pressure
The influence of pressure alters the manner in which gases, solids, and liquids dissolve within a solution. Altering the pressure has little impact on the solubility of solid and liquid materials. Conversely, an increase in gas pressure allows for better dissolution.
Polymorphs
Almost every crystal has the ability to take on various shapes or structures. Different forms of the same substance often exhibit variations in characteristics such as melting point. The ability of a solid to dissolve in a liquid is related to its melting point. Various polymorphic forms of the same substance can exhibit different solubility behaviors, despite being composed of the identical material. Typically, the disparity in the dissolving rates of a crystal relative to its various forms is approximately two to three times.The explanation lies in the slight differences in energy between these various forms [33].
Polarity
In basic terms, non-polar substances blend effectively with liquids that are also non-polar and lack electrical charges. In a similar way, charged substances (polar solutes) tend to blend effectively with liquids that possess similar charges (polar solvents). This is due to the fact that solute molecules possess both a positive and a negative component. Additionally, when a molecule has both a positive and a negative side, only the positive portion of the liquid will attract the negative side of the solute being dissolved. The type of attraction that occurs between molecules is known as a dipole-dipole interaction. Due to London dispersion forces, the positive regions of the solute molecule attract the negative electrons from the solvent molecules. The characteristics of both the solute and solvent play a crucial role in the solubility of the solute [34].
7. TECHNIQUES OF SOLUBILITY ENHANCEMENT
A substance's solubility and its ease of passing through barriers are significant determinants of how effectively drugs administered orally are absorbed by the body. Various techniques can be employed to modify or enhance these processes [35].
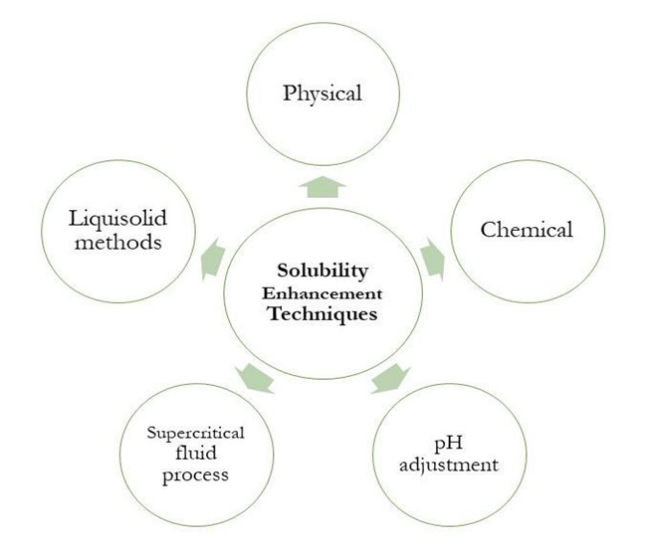
Figure 2: Broad Classification of Solubility Enhancement Techniques.
There are both traditional and modern techniques to enhance the solubility of substances [36].
7.1 Physical Modification
7.1.1 Particle Size Reduction
7.1.2 Modification of Crystal Habit
7.1.3 Drug Dispersion in Carrier
7.1.4 Solubilization by Surfactants
7.1.5 Complexation
7.2 Chemical modification
7.2.1 Hydrotrophy
7.2.2 Co-solvency
7.2.3 Nanotechnology
7.2.4 Salt Formation
7.3 pH adjustment
7.4 Supercritical fluid process
7.5 Liquisolid methods
7.1 Physical Modification
7.1.1 Particle Size Reduction
The effectiveness of a poorly soluble drug in the body is influenced by the size of its particles. Reducing the size of a particle increases its surface area. This enhances the substance's interaction with the liquid it's combined with, improving its dissolving ability. This is particularly beneficial for medications that have poor solubility. This provides numerous ways of formulation approaches as well as delivery technologies [37]. Various techniques such as micronization, nanosuspension, sonocrystallization, and spray drying can effectively decrease particle size. Additionally, there are various novel methods, such as solid self-emulsifying drug delivery systems, polymeric micelles, freeze-dried liposomes, and solid lipid nanoparticles.A variety of theories have been presented to elucidate the approaches for miniaturizing items [38].
Griffith theory: The force required is determined by the length of the crack and the focal point of stress at its tip.
Kick's law: When you diminish a specific quantity of material using a constant ratio, the reduction in size will consistently be the same, regardless of the initial size.
Rittinger's law: As particles decrease in size, their surface area increases significantly.
Bond's law: The size of a smaller particle corresponds to the square root of the original particle's size.
Micronization:
The Micronization technique enhances the rate at which drugs dissolve by reducing particle size, thereby increasing their surface area. However, the quantity that can dissolve does not change.Enhancing the surface area of these substances, thereby reducing their particle size, aids in accelerating their dissolution rate.
The techniques were applied to progesterone, fenofibrate, griseofulvin, spironolactone, and diosmin. These medications are more effective in the body, have improved absorption in the stomach, and provide better treatment outcomes for conditions [39].
Nanosuspension:
This technology is employed for medications that have poor solubility in either water or oil. Nanosuspension refers to a system characterized by nano size particles that are suspended in a aqueous solution. A surfactant is employed to maintain the stability of small drug particles used in injections, inhalation, oral administration, and skin applications. The solid particles within a nano suspension are generally smaller than one micron in size. The particles typically range in size from 200 to 600 nanometers. This approach was implemented for the compounds tarazepide, atovaquone, amphotericin, paclitaxel, and buparvaquone. Nanosuspensions can be produced through various methods such as Nanocrystals, DissoCubes, Nanopore, and Nano Edge [40].
7.1.2 Modification of Crystal Habit
Polymorphs
Polymorphism refers to the phenomenon where a solid can exist in two or more distinct crystal structures that are organized in various configurations. Polymorphs refer to various forms of crystal structures.
Pseudo polymorphs
Various crystalline forms of medications consist of identical chemicals, yet they exhibit distinct physical characteristics like melting temperature, texture, density, solubility in liquids, and stability. In a comparable manner, the amorphous form of a drug outperforms the crystalline form.The larger surface area and the significant energy involved are the reasons for this [41].
Order of different solid form of drugs
Amorphous > Metastable polymorphs > Stable polymorphs.
7.1.3 Drug Dispersion in Carrier
Solid solution: Two crystal solids are combined to form a new crystalline substance. Two substances are blended together in a homogeneous one-phase system to form a mixed crystal.In comparison to a basic enteric system, its dissolution rate is considerably higher [42].
Solid dispersion: The concept of solid dispersion was introduced by Sekiguchi and Obi. Solid dispersion is a valuable technique in pharmaceuticals that enhances the solubility, absorption, and efficacy of medications. "Solid dispersion" refers to a solid product characterized by the presence of both hydrophilic components and hydrophobic drugs. Widely utilized water-soluble carriers consist of polyethylene glycol, polyvinyl pyrrolidone, and Plasdone-S630. Solid mixtures are frequently produced using surfactants [43,44].
Eg. Common surfactants such as Myrj-52, Tween-80, sodium lauryl sulfate, Pluronic-F68, and docusate sodium serve different roles in formulations. This technique was applied to dissolve halofantrine, celecoxib, and ritonavir. Methods to create solid mixtures of hydrophobic drugs that improve their solubility in water include the following.
Fusion process: During this procedure, the carrier is subjected to heat until it changes to a liquid state. After that, the drug is introduced and thoroughly mixed in during the stirring process. Subsequently, the mixture is allowed to cool in order to achieve an even distribution of the drug throughout the matrix [45].
Solvent evaporation method: A well-formulated organic liquid blends the carrier with the active ingredient. The solvent is evaporated by applying temperature under vacuum conditions, resulting in a solid residue[46].
Frequently used solvents are chloroform, ethanol, or a mixture consisting of dichloromethane and ethanol.
7.1.4 Solubilization by Surfactants
Microemulsion: A microemulsion is a homogenous mixture that is both clear and stable in appearance. It includes oil, a unique surfactant, and a hydrophilic solvent that aids in dissolving a drug that is not readily soluble in water. When selecting a surfactant, it's important to take into account HLB and safety considerations.
When the mixtures come into contact with water, they effortlessly combine to form a clear liquid containing small, uniform droplets of oil.Rephrase These droplets contain medications that are poorly soluble in water, along with proteins suitable for oral administration, injections, or intravenous delivery. An oil-in-water (o/w) microemulsion is considered the most effective type of mixture. This blend is formulated to aid in dissolving substances that are not easily mixed with water by integrating them into the oil component [47].
Self-micro emulsifying drug delivery system
Self-emulsifying and self-microemulsifying systems generate an emulsion directly within the digestive tract. The self-emulsifying drug delivery system (SEDDS) is formed by blending oil, a soap-like compound, another soap-like material, one or more hydrophilic solvents, and a co-solvent to produce a transparent, homogeneous solution [48].
When it interacts with water in the digestive system, it independently forms small oil-in-water emulsions without needing any external water. This procedure aids in increasing the dissolution and absorption efficiency of particular oily pharmaceuticals. The process of mixing liquids is facilitated by water's ability to easily infiltrate the different solid or gel layers present on the surface of a droplet. One benefit of SEDDS for production is that they easily form when their ingredients are mixed together gently and they stay stable over time. The system faces challenges include chemical instabilities of drugs and high surfactant concentrations. Self-emulsifying products contain a significant concentration of surfactant (30-60%), which may cause irritation to the digestive tract. Due to their liquid nature, most self-emulsifying products are typically encapsulated in soft or hard gelatin capsules filled with lipid. It’s essential to consider how the capsule shell and the liquid within collaborate to prevent the contents from drying out or migrating into the shell [49].
An illustration of a self-microemulsifying drug delivery system (SMEDDS) is Neoral®. Depending on the dosage, the body may utilize 174% to 239% more cyclosporine A from Neoral® compared to Sandimmune®, the original marketed formulation [50]. The absorption of drugs from emulsion mixtures is significantly influenced by the size of the emulsion droplets, small droplets enhance the drug's presence in the bloodstream since they can be absorbed directly by the lymphatic system. Due to the high concentration of surfactants in SMEDDS, they are intended solely for oral use, and prolonged consumption may lead to diarrhoea [51].
Table 2. Marketed formulation using SEDDS drug delivery
|
Trade Name |
Drug Used |
Dosage Form |
Company |
References |
|
Neoral |
Cyclosporin |
Soft Gelatin |
Novartis |
[52] |
|
Norvir |
Ritonavir |
Capsule Soft Gelatin |
Abbott Laboratories |
[53] |
|
Fortovase |
Saquinavir |
Soft Gelatin Capsule |
Hoffmann roche |
[54] |
7.1.5 Complexation
Cyclodextrins have been utilized alongside drugs to enhance their solubility in water and maintain stability. In pharmaceutical applications, the most frequently utilized forms of β-cyclodextrin are those that have improved solubility in water. With a molecular weight greater than 1000 Da, cyclodextrins are considered large molecules that probably have difficulty to penetrate the skin. Observations have shown that skin permeability can fluctuate based on the presence of cyclodextrin. Besides their role in promoting better dissolution, CDs contribute to the increased permeability of membranes and the stability of substances.The presence of cyclodextrins enhances the ability of substances to move across biological membranes.CDs can aid in improving the permeability of medicines in systems designed for pulmonary drug delivery [55].
Table 3. Marketed formulation using complexation with cyclodextrins technique
|
Trade Name |
Drug Used |
Dosage Form |
Company |
References |
|
Nitropen |
Nitroglycerine/ βCD |
Sublingual tablet |
Nippon Kayaku |
[56] |
|
Nimedex |
Nimesulid/ βCD Omeprazole/ βCD |
Oral Sachet |
Novartis |
[57] |
|
Omebeta |
Nimesulid/ βCD Omeprazole/ βCD |
Tablet |
Betapharm |
[58] |
7.2 Chemical Modification
7.2.1. Hydrotrophy
Hydrotropy refers to the enhancement of a substance's solubility in water achieved by incorporating a large amount of an alternative substance [59]. A robust aqueous mixture containing sodium benzoate, sodium salicylate, urea, nicotinamide, sodium citrate, and sodium acetate has been observed to enhance the solubility of various poorly water-soluble drugs. Numerous substances have been identified that exhibit hydrotropic properties.
Some instances of this category are alcohols like resorcinol, pyrogallol, catechol, naphthalene, and salicylates. Substances referred to as alkaloids, such as caffeine and nicotine, are also present. Furthermore, ionic surfactants include compounds like diacids, sodium dodecyl sulfate (SDS), and dodecylated oxidized benzene [60]. Saquinavir, a medication, exhibits improved solubility in specific solutions. Its solubility increased by 473 times when combined with ascorbic acid, 462 times with nicotinamide, 49 times with resorcinol, and 52 times with dimethyl urea [61].
7.2.2 Co-solvency
Co-solvency refers to the use of one or more liquid solvents that effectively blend to improve the solubility of medications. Incorporating a co-solvent can enhance the solubility of the solution and improve its mixing capabilities. The co-solvent significantly improved the dissolution of the drug, making it nearly a thousand times more effective than the standard medications [62]. A co-solvent technique may be ideal for poorly soluble, lipophillic, or highly crystalline molecules, as long as they readily dissolve in the combination of solvents used. Numerous co-solvents are considered safe and effective for dissolving nonpolar drugs, making them primarily suitable for injection formulations.
To minimize the solvent levels before giving the medication, it may be necessary for parenteral formulations to incorporate water or be mixed with a liquid containing water. Weakly soluble substances can be made to dissolve more effectively by incorporating additional solvents and modifying the pH [63]. The application of co-solvents is a beneficial strategy for dissolving drugs that have low solubility. The most frequently used safe liquids in injections include propylene glycol, ethanol, glycerin, and polyethylene glycol. Dimethyl sulfoxide (DMSO) and dimethylacetamide (DMA) are frequently utilized as solvents for poorly soluble medications, as they are effective at dissolving a wide range of such drugs and are considered relatively safe [64].
7.2.3 Nanotechnology
Nanotechnology involves exploring and utilizing materials and structures that measure approximately 100 nanometers or less. Micronization, which involves creating small particles, can enhance the absorption of certain new medications in the body. This is because the micronized particles still have a limited surface area for dissolving. Thus, the next phase was to generate smaller particles referred to as nanonization [65].
7.2.4 Salt Formation
By employing salt- generation techniques, we can enhance the solubility of drugs.This approach is employed to observe the effects of various drugs or chemicals. The ionization of a medicine results in the formation of salt. It is effective in both injectable and liquid dose forms, as well as in solid dosage forms. Between 1995 and 2006, the FDA authorized the sale of over 300 new chemical products, with 120 of them being salt forms. Furthermore, hydrochloric acid was utilized in the preparation of 54 out of the 101 approved salt forms for basic drugs, indicating that hydrochloride salt was the most frequently used variant.
Changes in pH alter the solubility of acidic or basic drugs in water, thereby influencing their potential to form acceptable salts. Various methods are employed to alter the drug's stability, its bioavailability, purity, and manufacturability. For numerous years, pharmaceutical formulations have utilized salt forms of medications with poor solubility to facilitate better dissolution [66]. Examples of substances include Aspirin, Barbiturates, and Theophylline. Progesterone is a steroid that is not soluble in water but can be dissolved in peanut oil. is a commercially accessible example of this method [67].
7.3 pH adjustment
Adjusting the pH of water could improve the solubility of a drug that poorly dissolves in it. In order to evaluate the dissolving capability of a substance with this method, it's crucial to consider the solution's stability against pH fluctuations and the safety of the selected pH level. Some substances that dissolve in solutions can elevate the pH, thereby making it more alkaline. At this point, weakly acidic medications are more readily soluble. Likewise, components that increase the alkalinity of the solution can aid in the improved solubility of weakly basic medications [68].
7.4 Supercritical fluid process
Carbon dioxide, when utilized as a supercritical fluid at its critical point, can aid in dissolving substances that do not readily evaporate. It is safe, good for the environment, and affordable. A supercritical fluid (SCF) is a distinct phase that occurs when a substance exceeds its critical temperature and pressure. SCFs possess beneficial properties for product formulation, as they exist in a state between a complete liquid and a gas.
Furthermore, the density and flow characteristics of a substance—such as its viscosity and diffusivity—along with various physical properties like dielectric constant and polarity, can vary significantly with minor alterations in temperature, pressure, or both when near critical points. The unique applications of supercritical fluids (SCFs) that have been utilized in the food industry for years are now finding their way into the medical field. Frequently used supercritical solvents include carbon dioxide, nitrous oxide, ethylene, propylene, propane, n-pentane, ethanol, ammonia, and water.
Numerous methods have been developed to enhance Supercritical Fluid (SCF) processing in order to address particular issues. Among these methods are the employment of compressed antisolvents for precipitation (PCA), the rapid expansion of supercritical solutions, recrystallization using gas antisolvents, the addition of polymers featuring active materials, the use of compressed fluid antisolvents, improvements in dispersion with supercritical fluids (SEDS), the utilization of aerosol supercritical extraction systems (ASES), and the implementation of supercritical antisolvent procedures (SAS) [69].
7.5 Liquisolid methods
When a liquid containing a drug is introduced to a substance with porous surface and fibers, such as cellulose, two things occur: initially, the liquid is absorbed by the particles of the material. When the inside of the particles reaches capacity, the liquid adheres to both their inner and outer surfaces. Certain powders can be mixed with a liquid medicine to convert it into a dry powder that is non-sticky, easily portable, and compressible. Coating materials are made up of micro crystalline and amorphous forms of cellulose and silica powders [70,71].
CONCLUSION
The effectiveness of orally administered medications heavily depends on their solubility, as this influences their absorption into the bloodstream. A drug's solubility plays a crucial role in its formulation and overall effectiveness. The efficiency of absorption of weakly water-soluble drugs in the oral cavity is reliant on the drug's ability to dissolve effectively. The ability of a substance to dissolve is a crucial consideration in the formulation of various types of medication. The techniques described earlier can be utilized individually or in combination to enhance the solubility of molecules or difficult-to-dissolve medications. Numerous medications are difficult for the body to absorb due to poor solubility, so it's essential to explore methods to enhance their dissolution. The technique to enhance a substance's solubility is influenced by the drug's properties, including its chemical composition, physical state, and pharmacokinetic behaviour, and so on. Numerous techniques, similar to those discussed earlier, are now available to enhance the solubility of poorly soluble medications.
REFERENCES


Zoobi Tahir, Deepanshi Tyagi, Saurabh Mishra, Vimal Kumar Singh, A Comprehensive Review on Solubility Enhancement Techniques for Poorly Water-Soluble Drugs, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 839-855. https://doi.org/10.5281/zenodo.18494205
 10.5281/zenodo.18494205
10.5281/zenodo.18494205