
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,3,4,5,6,7,8,9,10 Matoshri College of Pharmacy, Eklahare, Nashik
2 K. V. N. Naik College of Pharmacy Canada Corner Nashik
Chromatography is an indispensable analytical tool in the pharmaceutical industry, playing a critical role in ensuring drug safety, efficacy, and quality through the separation, identification, and quantification of individual components within complex mixtures. The evolution of chromatographic techniques has significantly enhanced the precision, reliability, and throughput of pharmaceutical analyses. Among the most widely utilized methods are High-Performance Liquid Chromatography (HPLC), Ultra-High Performance Liquid Chromatography (UHPLC), Gas Chromatography (GC), and Supercritical Fluid Chromatography (SFC), each offering unique advantages based on their underlying principles, instrumentation, and compatibility with various analyses and matrices. This paper presents a comprehensive overview of modern chromatographic methods, delving into their theoretical foundations, operational mechanisms, and key instrumentation components, such as mobile and stationary phases, detectors, and data processing systems. HPLC remains a cornerstone in pharmaceutical quality control and drug development due to its versatility and high resolution, while UHPLC represents a more recent advancement, providing increased speed and sensitivity through the use of smaller particle sizes and higher pressures. GC, with its applicability to volatile and semi-volatile compounds, continues to be a preferred method for the analysis of gases and organic solvents, whereas SFC is gaining prominence for its eco-friendliness and efficiency in separating chiral and thermally labile compounds. Furthermore, this paper explores the stepwise formulation procedures for developing and validating chromatographic analytical methods in compliance with regulatory guidelines issued by agencies such as the International Council for Harmonization (ICH), United States Pharmacopeia (USP), and European Medicines Agency (EMA). Emphasis is placed on method development strategies, selection of analytical parameters, and optimization techniques to achieve robust and reproducible results. Key evaluation parameters—including system suitability, specificity, linearity, accuracy, precision, limit of detection (LOD), limit of quantification (LOQ), robustness, and ruggedness—are discussed in detail to underscore the critical aspects of method validation. The regulatory significance of chromatographic methods is also examined, highlighting their essential role in ensuring compliance with Good Manufacturing Practices (GMP), quality assurance, and product lifecycle management. Analytical data generated through validated chromatographic methods serve as pivotal evidence for regulatory submissions, stability studies, and batch release protocols. In conclusion, chromatography remains at the forefront of pharmaceutical analysis, with continual advancements driving improvements in analytical performance and regulatory compliance. The integration of modern chromatographic technologies into pharmaceutical workflows enhances the ability to develop safer, more effective drugs, thereby supporting public health and global therapeutic innovation.
Pharmaceutical analysis is a cornerstone of modern drug development and quality assurance, serving as the scientific discipline responsible for verifying the identity, strength, purity, and composition of pharmaceutical products. This essential process encompasses both qualitative and quantitative evaluations, ensuring that pharmaceutical compounds meet the stringent standards established by global regulatory authorities such as the United States Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). In an era of increasing complexity in drug formulations and growing public health demands, pharmaceutical analysis has assumed an even more vital role in safeguarding patient safety, optimizing therapeutic outcomes, and supporting the integrity of pharmaceutical manufacturing. Among the wide array of analytical techniques employed in pharmaceutical analysis, chromatographic methods have emerged as particularly indispensable. These techniques allow for the precise separation, identification, and quantification of active pharmaceutical ingredients (APIs), excipients, degradation products, impurities, and metabolites present in complex mixtures. Chromatography is especially valued for its high resolution, sensitivity, selectivity, and reproducibility, making it suitable for a broad range of substances, from small organic molecules to large biomolecules. According to Sharma (2022), the consistent accuracy and versatility of chromatographic techniques have made them the analytical methods of choice across various stages of the drug lifecycle—from initial discovery and formulation development to manufacturing control and post-market surveillance. The origins of chromatography date back to the early 20th century, but its application in the pharmaceutical sciences has expanded dramatically with the advent of modern techniques. Key developments such as High-Performance Liquid Chromatography (HPLC), Ultra-High Performance Liquid Chromatography (UHPLC), Gas Chromatography (GC), and more recently, Supercritical Fluid Chromatography (SFC) have revolutionized pharmaceutical analysis. These innovations have not only enhanced the speed, resolution, and sensitivity of analytical procedures but have also facilitated automation, miniaturization, and compliance with evolving environmental and regulatory standards. Each chromatographic technique offers distinct advantages depending on the chemical nature of the analytes and the objectives of the analysis. For example, HPLC is widely used for stability testing and content uniformity, GC is preferred for volatile compounds, and SFC is gaining popularity for chiral separations and green chemistry applications. The success of chromatographic analysis in pharmaceutical settings is also deeply intertwined with method development and validation processes. Analytical method development involves selecting suitable conditions—such as mobile and stationary phases, flow rate, detection wavelength, and sample preparation—to achieve optimal separation and quantification of target compounds. Validation, as outlined in ICH guidelines (e.g., ICH Q2(R1)), ensures that the developed method meets key performance criteria, including specificity, linearity, accuracy, precision, detection limit, quantitation limit, robustness, and system suitability. A validated method is not only a regulatory requirement but also a scientific guarantee of the reliability and consistency of analytical results. Moreover, chromatographic methods play a pivotal role in addressing various analytical challenges associated with new drug entities, combination products, controlled-release formulations, and biologics. As pharmaceutical formulations become more sophisticated, chromatographic methods are increasingly used in the characterization of drug-excipient interactions, identification of trace-level impurities, enantiomeric purity testing, and pharmacokinetic studies. In the context of regulatory compliance and quality management systems, these methods support the implementation of Good Manufacturing Practices (GMP) and Quality by Design (QbD) principles, thereby ensuring continuous process verification and lifecycle management.This paper aims to provide a comprehensive overview of chromatography in pharmaceutical analysis, focusing on its principles, types, instrumentation, method development strategies, validation procedures, evaluation parameters, and regulatory significance. Through this examination, the paper highlights how chromatographic techniques have transformed pharmaceutical quality control and contributed significantly to the advancement of pharmaceutical sciences and healthcare deliver[1].
2. Principles of Chromatography
Chromatography is a powerful analytical technique that relies on the fundamental principle of differential partitioning or differential migration of components in a mixture between two distinct phases: the stationary phase and the mobile phase. This differential distribution of solutes leads to their separation, enabling their individual detection, identification, and quantification. First introduced in the early 20th century by the Russian botanist Mikhail Tswett, chromatography has evolved into a multifaceted tool that underpins much of modern pharmaceutical, environmental, and biochemical analysis (Ahuja, 2005). At its core, the separation mechanism in chromatography is governed by the relative affinities of individual analytes toward the mobile and stationary phases. The mobile phase is a fluid (either a gas or a liquid) that carries the mixture of compounds through the stationary phase, which is a solid or a liquid immobilized on a solid support. As the sample traverses the chromatographic system, each component interacts with the two phases based on its chemical and physical properties, such as polarity, molecular weight, volatility, or hydrophobicity. Components that interact more strongly with the stationary phase move more slowly, while those with a higher affinity for the mobile phase elute faster, resulting in a temporal and spatial separation. For example, in liquid chromatography, a polar stationary phase and a non-polar mobile phase (in normal-phase LC), or vice versa (in reversed-phase LC), can be selected depending on the nature of the analytes. In gas chromatography, the volatility and thermal stability of compounds determine their retention times, as the mobile phase is an inert gas and the separation occurs within a heated column. In all types of chromatography, the extent of separation is measured in terms of retention time (tR)—the time it takes for each component to reach the detector—and retention factor (k'), which quantifies the degree of retention of each solute relative to a non-retained compound. The mechanism of separation can further involve several types of interactions, including:
These interactions can be fine-tuned by adjusting parameters such as the mobile phase composition, pH, flow rate, temperature, and nature of the stationary phase. This tunability makes chromatography exceptionally versatile and adaptable to a wide range of analytical challenges. In the pharmaceutical sciences, understanding the partitioning behavior of analytes is crucial for method development and optimization. Accurate separation allows for the detection of impurities, identification of degradation products, and quantification of active pharmaceutical ingredients (APIs), all of which are essential for ensuring drug quality and safety. Furthermore, the use of detectors such as UV-Vis, fluorescence, mass spectrometry, and refractive index extends the capabilities of chromatographic techniques, allowing the analyst to gain insight into the identity and concentration of analytes at trace levels. Mathematically, chromatographic separation is often described by concepts such as theoretical plates (N), which indicate column efficiency; selectivity (α), which measures the separation between peaks; and resolution (Rs), which quantifies how well two components are separated. An optimal chromatographic system is one where all components of interest elute as sharp, well-resolved peaks, allowing accurate quantitative and qualitative assessments. In conclusion, the principle of chromatography—differential partitioning between a mobile and stationary phase—forms the scientific basis for one of the most powerful and widely used separation techniques in analytical chemistry. As analytical demands grow increasingly complex in the pharmaceutical and biomedical sciences, the understanding and application of chromatographic principles remain foundational to achieving precise, reliable, and regulatory-compliant results (Ahuja, 2005)[2].
Table: Summary of Chromatographic Techniques and Their Key Characteristics
|
Chromatography Type |
Mechanism |
Stationary Phase |
Mobile Phase |
Main Interaction Type |
Common Applications |
|
Adsorption Chromatography |
Adsorption |
Solid (e.g., silica, alumina) |
Liquid or gas |
Surface adsorption |
Separation of small organic molecules, pigments, drugs |
|
Partition Chromatography |
Partition |
Liquid coated on inert solid support |
Liquid or gas |
Solute partitioning between phases |
Separation of non-polar and polar solutes, drug metabolites |
|
Ion-Exchange Chromatography |
Electrostatic attraction |
Resin with charged functional groups |
Aqueous solution (buffer) |
Ionic (charge-based) interactions |
Separation of amino acids, peptides, proteins, nucleotides |
|
Size-Exclusion Chromatography (SEC) |
Size-based sieving |
Porous gel (e.g., dextran, agarose, polyacrylamide) |
Aqueous or organic solvents |
Molecular size exclusion |
Protein, polymer, and macromolecule separation and molecular weight analysis |
|
Affinity Chromatography |
Biological interaction |
Matrix with immobilized ligand (e.g., antibody) |
Aqueous buffer with elution agent |
Specific ligand-analyte binding |
Purification of proteins, enzymes, monoclonal antibodies |
|
Gas Chromatography (GC) |
Partition/Volatility |
Liquid film on capillary inner wall or solid support |
Inert gas (He, N?, H?) |
Volatility and partitioning |
Analysis of volatile and semi-volatile compounds, residual solvents |
|
High-Performance Liquid Chromatography (HPLC) |
Partition/Adsorption |
Silica or modified silica (C18, C8, CN, etc.) |
Polar or non-polar liquids (acetonitrile, methanol, water) |
Hydrophobic/hydrophilic interactions |
Assay of drugs, impurities, degradation products |
|
Ultra-High Performance Liquid Chromatography (UHPLC) |
Partition |
Sub-2 µm particles (C18, C8, etc.) |
Similar to HPLC but under higher pressure |
Similar to HPLC |
Faster, high-resolution separation of pharmaceuticals |
|
Supercritical Fluid Chromatography (SFC) |
Partition |
Chiral or non-chiral packed columns |
Supercritical CO? + organic modifier |
Polarity, stereochemistry |
Enantiomer separation, analysis of lipophilic and thermally labile drugs |
3. Advanced Chromatographic Techniques
3.1 High-Performance Liquid Chromatography (HPLC)
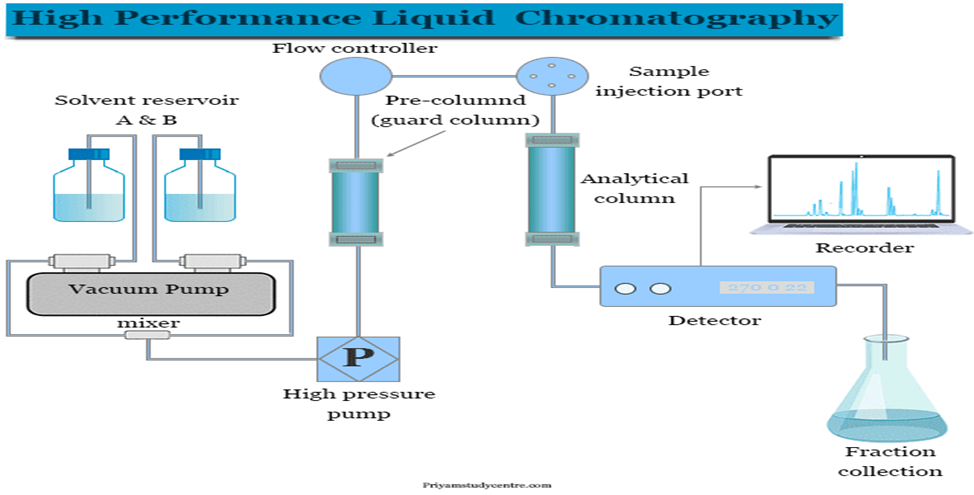
High-Performance Liquid Chromatography (HPLC) is one of the most widely employed analytical techniques in pharmaceutical analysis, particularly well-suited for the separation, identification, and quantification of non-volatile and thermally unstable compounds. Unlike gas chromatography (GC), which requires analytes to be volatile and thermally stable due to the elevated temperatures involved in the process, HPLC operates at ambient or moderately elevated temperatures, making it ideal for a wide range of drug substances, biologically active molecules, and excipients that would degrade or decompose under the conditions required for GC analysis (Kazakevich & Lobrutto, 2007). The fundamental principle of HPLC lies in the partitioning of analytes between a liquid mobile phase and a solid or liquid stationary phase, typically packed into a stainless steel column. The technique functions under high pressure, usually in the range of 4000 to 6000 psi (and up to 10,000+ psi for UHPLC systems), which forces the mobile phase through the tightly packed column and enhances the efficiency and resolution of the separation. The increased pressure allows for the use of columns filled with small particle sizes (3–5 µm for conventional HPLC and sub-2 µm for UHPLC), which provide greater surface area and improved interactions between analytes and the stationary phase. HPLC can be operated in various modes, including reversed-phase (RP-HPLC), normal-phase, ion-exchange, and size-exclusion chromatography, depending on the polarity, charge, and size of the analytes. Reversed-phase HPLC, which employs a non-polar stationary phase (commonly C18) and a polar mobile phase (typically water mixed with organic solvents like methanol or acetonitrile), is the most commonly used configuration in pharmaceutical analysis due to its broad applicability and robust performance. One of the critical advantages of HPLC is its versatility in detection methods, which significantly contributes to its analytical power. The most commonly used detectors in HPLC include:
The ability to pair HPLC with various detectors enables analysts to tailor the system according to the chemical nature and concentration of the analytes under investigation. For example, UV detection is often sufficient for routine pharmaceutical QC testing, while MS detection is favored in bioanalytical studies, impurity profiling, and stability-indicating methods. In addition to its analytical capabilities, HPLC is integral to method development, validation, and regulatory submissions. Regulatory authorities, including the FDA and EMA, often require that HPLC methods be used to demonstrate the identity, strength, purity, and stability of pharmaceutical products. These methods must be developed and validated according to ICH guidelines (e.g., Q2(R1)), ensuring that the analytical procedure is specific, accurate, precise, linear, robust, and reproducible. HPLC also plays a vital role in pharmacokinetic studies, bioequivalence testing, drug dissolution testing, and batch release testing. In formulation development, HPLC aids in determining the compatibility of active ingredients with excipients and in monitoring degradation products under stress conditions. It is also widely used for the quantification of residual solvents and impurities, both of which are crucial for ensuring product safety and compliance with regulatory limits. In summary, High-Performance Liquid Chromatography is a cornerstone analytical technique in pharmaceutical science, offering high-resolution, reproducibility, and the ability to handle a diverse range of non-volatile and thermally labile substances. Its compatibility with a wide array of detectors, coupled with its robustness and regulatory acceptability, ensures that HPLC will continue to be a central tool in drug development, quality control, and regulatory compliance (Kazakevich & Lobrutto, 2007)[3].
3.2 Ultra-High Performance Liquid Chromatography (UHPLC)
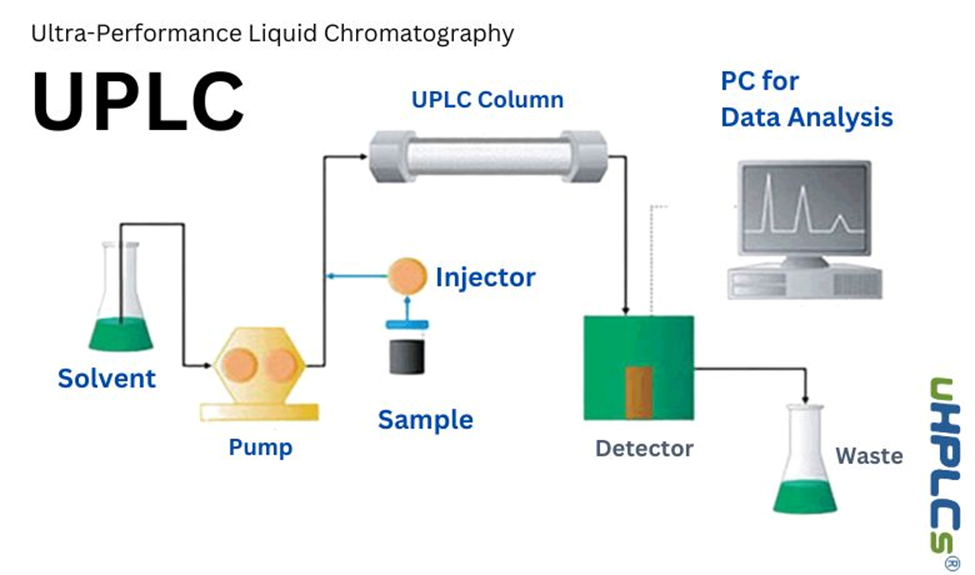
Ultra-High Performance Liquid Chromatography (UHPLC), sometimes referred to as UPLC (Ultra Performance Liquid Chromatography), represents a significant advancement over conventional High-Performance Liquid Chromatography (HPLC). It was developed in response to the growing demand for faster, more efficient, and more sensitive analytical techniques in the pharmaceutical and biomedical sciences. UHPLC distinguishes itself from traditional HPLC by utilizing columns packed with sub-2 µm particle size stationary phases, typically in the range of 1.7 µm, and by operating at elevated system pressures, often exceeding 15,000 psi (Swartz, 2005). These innovations collectively result in improved chromatographic resolution, higher sensitivity, and significantly reduced analysis times, making UHPLC a preferred tool in both research and quality control settings. The reduced particle size is a central feature of UHPLC technology. According to chromatographic theory, particularly the van Deemter equation, using smaller particles decreases band broadening, increases the number of theoretical plates, and ultimately improves the efficiency and resolution of the separation. This makes UHPLC particularly effective in analyzing complex mixtures where closely eluting compounds must be separated with high precision. In practical terms, this means that peaks are sharper, better resolved, and detected more accurately, which is critical when analyzing drug substances, impurities, degradation products, and metabolites. Another notable advantage of UHPLC is its capacity to accelerate analytical throughput. Due to the smaller particle sizes and higher pressures, UHPLC columns can be run at higher flow rates without compromising performance, enabling much faster run times compared to standard HPLC methods. What might take 30–40 minutes using conventional HPLC can often be accomplished in 5–10 minutes using UHPLC, making it invaluable in environments where large volumes of samples must be processed quickly, such as in bioequivalence studies, batch release testing, or stability studies. In terms of detection, UHPLC systems are highly versatile and can be interfaced with various detectors such as UV/Vis, Photodiode Array (PDA), fluorescence, and mass spectrometry (MS). The combination of UHPLC with MS, especially tandem mass spectrometry (UHPLC-MS/MS), enables unparalleled specificity and sensitivity for trace-level quantification and structural elucidation of compounds in complex matrices such as plasma, serum, or tissue homogenates. This makes it especially valuable in pharmacokinetic and toxicological studies, where accurate and sensitive detection of drugs and their metabolites is critical. The instrumental design of UHPLC systems has also evolved to support the demands of high-pressure operations. Components such as pumps, autosamplers, and columns are engineered to tolerate ultra-high pressures while minimizing system dead volume, which is essential for maintaining peak integrity and ensuring reproducibility. Additionally, advances in data processing software and automation have further enhanced the capabilities of UHPLC systems, enabling more efficient method development, real-time peak tracking, and sophisticated data analysis. From a regulatory perspective, UHPLC is recognized by major global health authorities including the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), provided that methods are validated appropriately. Analytical procedures developed using UHPLC must comply with ICH Q2(R1) guidelines for validation, demonstrating attributes such as specificity, accuracy, precision, linearity, limit of detection (LOD), limit of quantification (LOQ), robustness, and system suitability. When validated properly, UHPLC methods can be directly used for critical applications in new drug applications (NDAs), abbreviated new drug applications (ANDAs), clinical trials, and post-marketing quality control. Moreover, UHPLC aligns with the principles of green analytical chemistry due to its reduced consumption of organic solvents and shorter run times. This not only contributes to cost efficiency but also supports environmentally sustainable practices within pharmaceutical laboratories. The lower solvent use, combined with faster cycle times, allows laboratories to reduce waste and energy consumption, all while improving throughput and maintaining analytical rigor. In summary, UHPLC is a revolutionary enhancement of liquid chromatography that provides a powerful combination of high resolution, speed, sensitivity, and efficiency. Its ability to analyze a wide range of compounds, including non-volatile, thermally unstable, and low-concentration analytes, makes it indispensable in modern pharmaceutical analysis. As analytical demands continue to evolve with advances in drug development and regulatory expectations, UHPLC will undoubtedly remain a cornerstone technology in the analytical scientist’s toolkit (Swartz, 2005)[4].
3.3 Gas Chromatography (GC)
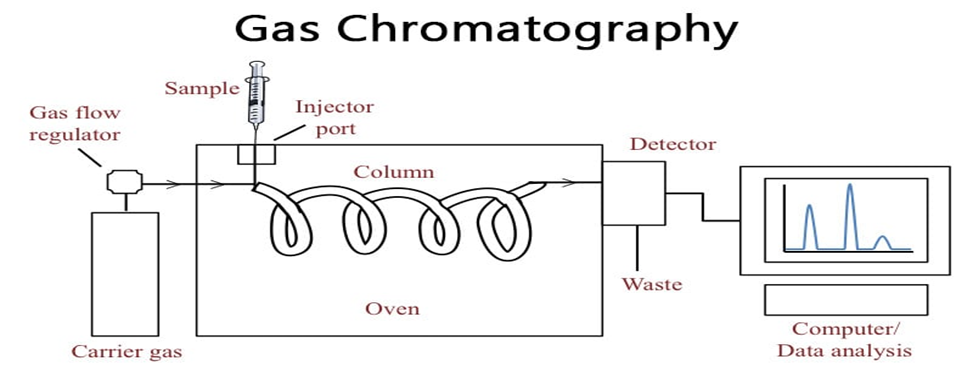
Gas Chromatography (GC) is one of the most widely utilized analytical techniques for the qualitative and quantitative analysis of volatile and semi-volatile compounds. Unlike liquid chromatography techniques, GC operates by using a gaseous mobile phase, typically an inert carrier gas such as helium, nitrogen, or hydrogen, to transport analytes through a stationary phase that is either coated onto the inner wall of a capillary column (in capillary GC) or packed into a steel or glass column (in packed GC) (McMaster, 2008). The technique is based on the principle that different compounds in a mixture will partition between the mobile and stationary phases at varying rates depending on their volatility and interactions with the stationary phase, thereby allowing for efficient separation. GC is particularly well-suited for substances that are volatile at elevated temperatures and thermally stable, making it ideal for the analysis of essential oils, organic solvents, hydrocarbons, pesticides, and residual solvents in pharmaceuticals. The process begins with the injection of a liquid or gaseous sample into a heated inlet port, where it is vaporized and swept into the column by the carrier gas. Inside the column, compounds are separated based on their boiling points and affinity for the stationary phase, which can be polar or non-polar depending on the analyte properties and desired selectivity. One of the major advantages of GC is its high resolution and reproducibility, enabling the accurate detection of trace components in complex mixtures. The method is especially valuable in pharmaceutical quality control for the detection of residual solvents, which must be quantified at very low levels in accordance with International Conference on Harmonisation (ICH) guidelines, particularly ICH Q3C, which governs acceptable limits for such impurities. Detection in GC is accomplished through highly sensitive and selective detectors, the most common of which are the Flame Ionization Detector (FID) and Mass Spectrometry (MS):
Other detectors such as the Electron Capture Detector (ECD), Thermal Conductivity Detector (TCD), and Nitrogen-Phosphorus Detector (NPD) are used based on the specific application, such as halogenated compounds, inorganic gases, or nitrogen/phosphorus-containing drugs.
In the pharmaceutical industry, GC finds significant application in:
GC methods are also integral to pharmacopoeial standards. Monographs in the United States Pharmacopeia (USP), European Pharmacopoeia (Ph. Eur.), and Japanese Pharmacopoeia (JP) frequently specify GC techniques for identifying and quantifying residual solvents and volatile impurities. As such, regulatory authorities expect validated GC methods during drug approval and post-marketing surveillance. GC method development involves careful optimization of parameters such as column type (capillary vs. packed), stationary phase polarity, carrier gas flow rate, oven temperature programming, and detector settings. Additionally, method validation according to ICH Q2(R1) guidelines ensures that the GC method meets the required standards of accuracy, precision, linearity, specificity, sensitivity (LOD & LOQ), and robustness. While GC is not suitable for the analysis of non-volatile or thermally labile compounds—where techniques like HPLC or UHPLC are preferred—it remains indispensable for volatile analytes due to its unmatched efficiency, speed, and sensitivity. In conclusion, Gas Chromatography stands as a cornerstone technique in analytical chemistry and pharmaceutical quality control, providing precise, rapid, and reliable analysis of volatile substances. Its adaptability with sensitive detectors like FID and MS further enhances its analytical capabilities, making GC an essential component of modern pharmaceutical analysis and regulatory compliance (McMaster, 2008)[5].
3.4 Supercritical Fluid Chromatography (SFC)
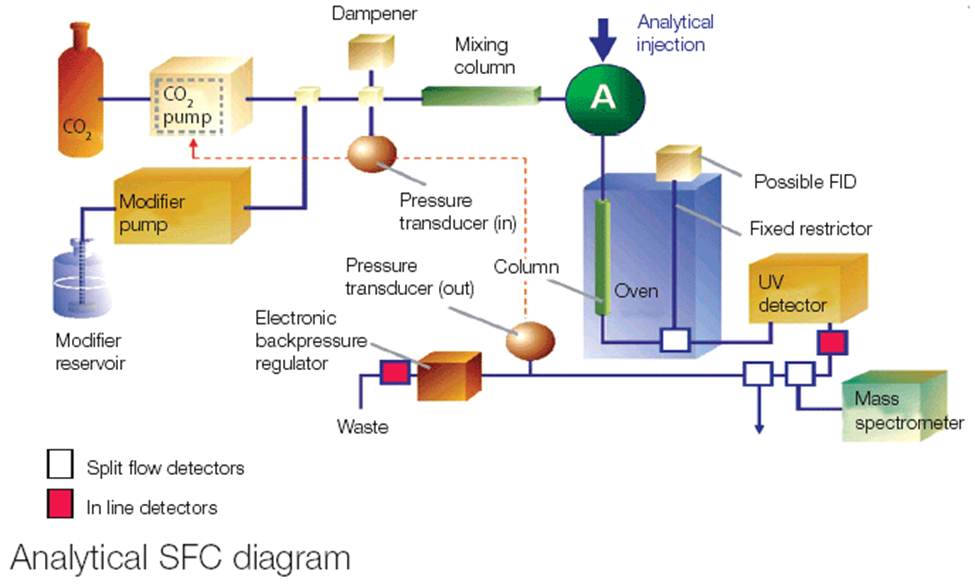
Supercritical Fluid Chromatography (SFC) is a highly versatile and increasingly utilized chromatographic technique that combines features of both gas chromatography (GC) and liquid chromatography (LC). SFC uses a supercritical fluid—most commonly carbon dioxide (CO?)—as the mobile phase, which behaves like a gas in terms of viscosity and like a liquid in terms of solvating power. This unique combination allows for rapid, efficient separations with high resolution and low solvent consumption (Berger, 2010). The supercritical state of CO? occurs when it is heated and pressurized above its critical temperature (31.1°C) and critical pressure (73.8 bar). In this state, CO? exhibits properties intermediate between gases and liquids: it diffuses more rapidly than liquids and has a lower viscosity, which enhances mass transfer and allows for faster analysis. These features make SFC particularly effective in separating compounds that are thermally sensitive or structurally complex, such as pharmaceutical intermediates, chiral drugs, and natural products. One of the most important applications of SFC is in chiral chromatography, where it has become the preferred technique for enantiomeric separations. Enantiomers—molecules that are mirror images of each other—can have profoundly different pharmacological effects, and regulatory agencies require that chiral drugs be thoroughly evaluated for enantiomeric purity. SFC offers exceptional enantioselectivity, often using chiral stationary phases such as polysaccharide- or protein-based materials, and provides faster separations than traditional normal-phase HPLC methods. SFC is also advantageous in the analysis of thermally labile compounds that might degrade under the high temperatures required in GC, and for compounds that are poorly soluble in water or require minimal organic solvent usage. This makes it particularly useful for the analysis of lipophilic drugs, steroids, esters, essential oils, and active pharmaceutical ingredients (APIs). It has also proven beneficial in the separation of isomers, purification of drug substances, and green chemistry applications, thanks to its reduced need for toxic and volatile organic solvents.
Instrumentation and Mobile Phase Composition
An SFC system closely resembles an HPLC system in design but incorporates modifications to handle pressurized CO? and maintain it in a supercritical state throughout the chromatographic run. The mobile phase is typically supercritical CO?, often modified with co-solvents (modifiers) such as methanol, ethanol, isopropanol, or acetonitrile to enhance solubility and modify selectivity. These co-solvents are added in varying percentages (usually 5–40%) depending on the polarity and complexity of the analytes. The system includes a CO? pump, a modifier pump, an auto-injector, a column oven, a back pressure regulator, and a detector—most commonly UV, PDA, FID, or MS. The back pressure regulator is a crucial component, ensuring that the CO? remains in a supercritical state throughout the column and the detector[6].
3.5 Hyphenated Techniques (LC-MS, GC-MS)
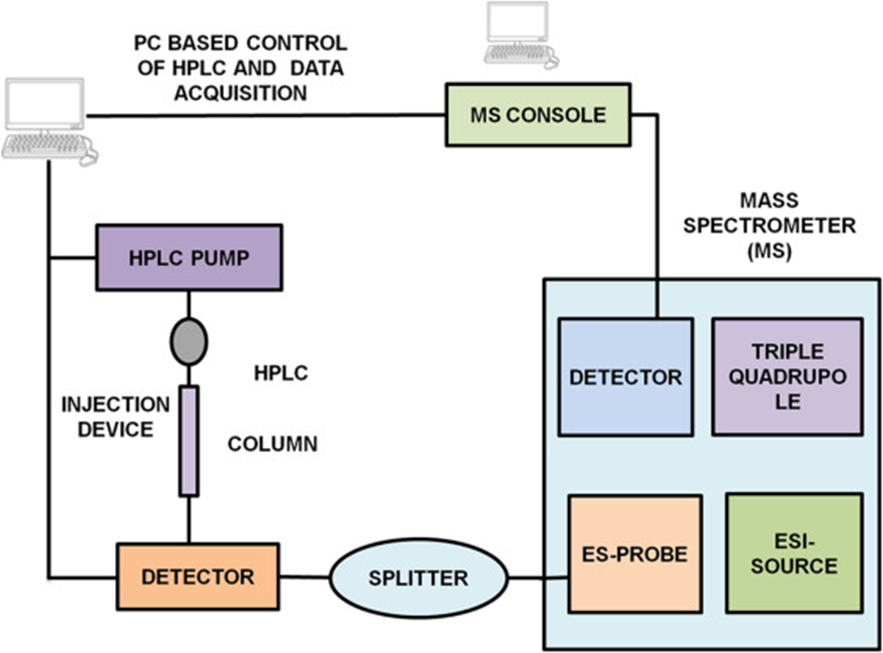
The combination of chromatographic separation techniques with mass spectrometric detection—commonly referred to as Chromatography-Mass Spectrometry (LC-MS or GC-MS)—has emerged as one of the most powerful and versatile tools in analytical chemistry, especially in pharmaceutical and biomedical research. This hybrid approach merges the high-resolution separation capabilities of chromatography with the molecular specificity, sensitivity, and structural elucidation potential of mass spectrometry (Niessen, 2006; Gross, 2011). Chromatography alone is highly effective for separating complex mixtures based on differences in chemical or physical properties such as polarity, charge, or size. However, its capability to identify and quantify trace-level components, particularly in complex biological matrices or multicomponent formulations, is significantly enhanced when coupled with a mass spectrometer. Mass spectrometry, on the other hand, provides precise molecular mass information, isotopic patterns, fragmentation data, and structural insights. When combined, the resulting system allows for the qualitative and quantitative analysis of components present at very low concentrations, often in the picogram to femtogram range. In LC-MS (Liquid Chromatography–Mass Spectrometry), high-performance liquid chromatography (HPLC) or ultra-high-performance liquid chromatography (UHPLC) is used to separate compounds, which are then introduced into the mass spectrometer via an interface such as an electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) source. These interfaces ionize analytes without fragmenting them, preserving their molecular integrity for accurate detection. The mass analyzer (such as a quadrupole, time-of-flight (TOF), ion trap, or Orbitrap) detects these ions based on their mass-to-charge ratio (m/z), allowing both qualitative identification and quantitative determination. GC-MS (Gas Chromatography–Mass Spectrometry) operates on a similar principle but is best suited for volatile, thermally stable compounds. Here, an inert gas (e.g., helium) transports analytes through a capillary GC column, which separates them based on volatility and interaction with the stationary phase. The eluted compounds are ionized using methods such as electron impact (EI) or chemical ionization (CI) before entering the mass analyzer. GC-MS is extensively used in the analysis of drugs of abuse, environmental pollutants, essential oils, residual solvents, and small organic molecules.
Advantages of Chromatography-Mass Spectrometry Coupling
Applications in the Pharmaceutical Industry
Challenges and Considerations
Despite its strengths, chromatography-mass spectrometry coupling requires careful method development, optimization of ionization conditions, and instrument maintenance. Matrix effects, ion suppression/enhancement, and complex data interpretation are common challenges. However, advancements in software algorithms, ionization sources, and high-resolution mass spectrometry have significantly improved robustness and usability[7,8].
Table: Comparative Overview of LC-MS and GC-MS in Pharmaceutical Analysis
|
Parameter |
LC-MS (Liquid Chromatography–Mass Spectrometry) |
GC-MS (Gas Chromatography–Mass Spectrometry) |
|
Type of Analytes |
Polar, non-volatile, thermally labile compounds |
Volatile, thermally stable compounds |
|
Mobile Phase |
Liquid (e.g., water, methanol, acetonitrile) |
Inert gas (e.g., helium, nitrogen) |
|
Stationary Phase |
Packed column with silica-based bonded phases |
Capillary columns with liquid stationary phase coated on inner walls |
|
Ionization Techniques |
Electrospray Ionization (ESI), APCI, APPI |
Electron Impact (EI), Chemical Ionization (CI) |
|
Detector/Analyzer |
Quadrupole, TOF, Orbitrap, Ion Trap |
Quadrupole, TOF, Ion Trap |
|
Sample Preparation |
Minimal; direct injection of biological fluids possible |
Requires derivatization for non-volatile samples |
|
Sensitivity |
High (picogram to femtogram levels) |
High (nanogram to picogram levels) |
|
Quantitative Accuracy |
Excellent with internal standards and MRM |
Very good with calibration curves |
|
Applications |
- Bioanalysis (e.g., plasma drug levels) - Metabolite profiling - Impurity testing - Pharmaceutical QC |
- Forensic toxicology - Environmental testing - Residual solvent analysis - Volatile impurity profiling |
|
Strengths |
- Suitable for thermolabile, high-molecular-weight compounds - Direct biological sample analysis |
- Excellent for volatile compounds - Robust and reproducible |
|
Limitations |
- Matrix effects (ion suppression) - Costly instrumentation |
- Requires derivatization for many pharmaceutical compounds |
4. Formulation Procedure for Analytical Method Development
Developing a chromatographic method involves a systematic approach:
1. Define Objective
E.g., identify, quantify, or monitor degradation.
2. Sample Preparation
Extraction, filtration, dilution depending on matrix.
3. Selection of Chromatographic Conditions
4. Trial Runs & Optimization
Change pH, organic ratio, gradient to improve separation.
5. System Suitability Testing
Parameters like retention time, resolution, and tailing factor checked before validation.
5. Evaluation Parameters (Method Validation as per ICH Q2(R1))
6. Applications in Pharmaceutical Analysis
Table: Applications of Chromatographic Techniques in Pharmaceutical Analysis
|
Application |
Most Suitable Chromatographic Technique(s) |
Purpose/Details |
|
1. Assay of Active Pharmaceutical Ingredients (APIs) |
HPLC, UHPLC |
Quantification of APIs in bulk drugs and finished dosage forms |
|
2. Impurity Profiling |
HPLC, LC-MS, GC-MS |
Detection and quantification of known and unknown impurities |
|
3. Bioequivalence and Pharmacokinetics |
LC-MS/MS |
Measurement of drug concentrations in plasma or blood over time |
|
4. Dissolution Testing |
HPLC, UHPLC |
Determines rate and extent of drug release from dosage forms |
|
5. Residual Solvent Analysis |
GC, GC-MS |
Detection and quantification of residual solvents in drug substances (per ICH Q3C) |
|
6. Chiral Separation |
SFC, Chiral HPLC |
Separation of enantiomers in chiral drug substances and intermediates |
|
7. Degradation Product Identification |
LC-MS, UHPLC, HPLC |
Identification of drug degradation pathways and stability-indicating methods |
7. Regulatory Importance of Chromatographic Methods in Pharmaceutical Analysis
Chromatographic techniques hold a critical position in the pharmaceutical industry, not only as essential tools for drug development and quality control but also as mandated analytical methodologies by regulatory authorities worldwide. Agencies such as the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), United States Pharmacopeia (USP), and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) have established stringent guidelines that require the use of validated chromatographic methods for the qualitative and quantitative analysis of pharmaceutical products (USP, 2023; ICH, 2006)[12]. The regulatory framework emphasizes the importance of chromatography in ensuring that drug products meet predefined standards of identity, purity, potency, and safety. These methods must undergo comprehensive validation to demonstrate their suitability for intended analytical purposes, including parameters such as specificity, accuracy, precision, linearity, limit of detection (LOD), limit of quantification (LOQ), robustness, and system suitability. This validation process is a prerequisite for submission dossiers during drug approval, ensuring that analytical data supporting product claims are reliable and reproducible. The FDA’s guidance documents explicitly state that chromatographic methods, such as HPLC or GC, should be developed and validated in accordance with the ICH Q2(R1) guidelines, which provide detailed recommendations on method validation. These regulatory expectations help in maintaining the integrity of pharmaceutical products throughout their lifecycle—from raw material testing and in-process control to final product release and stability studies. Additionally, chromatographic methods play a pivotal role in impurity profiling and degradation product identification, critical for evaluating drug safety and efficacy. Regulatory authorities mandate the establishment of stability-indicating methods that can separate and quantify the active pharmaceutical ingredient (API) and its potential impurities, including degradation products formed under stress conditions. These requirements help mitigate risks associated with toxic impurities and ensure that pharmaceutical formulations remain effective and safe during their shelf life. Moreover, residual solvent analysis by gas chromatography is a regulatory requirement under ICH Q3C, aimed at controlling solvents that may be harmful if present above certain limits. Similarly, chiral separations conducted using specialized chromatographic methods are required to characterize enantiomer purity for chiral drugs, addressing issues of efficacy and safety related to stereochemistry. In conclusion, the role of chromatography extends beyond analytical convenience—it is an indispensable component of regulatory compliance. The implementation of robust, validated chromatographic techniques ensures that pharmaceutical companies meet global standards, safeguarding patient health and enabling market access for new and generic drug products. This regulatory significance continues to drive innovation and refinement in chromatographic technologies and methodologies, reinforcing their status as pillars of pharmaceutical quality assurance (USP, 2023; ICH, 2006)[12].
8. Recent Advances and Future Trends in Chromatography
The field of chromatography is continuously evolving, driven by the need for faster, more efficient, and environmentally sustainable analytical methods. Recent technological advancements and emerging trends reflect the growing emphasis on green chemistry, miniaturization, automation, and data-driven optimization, all of which are reshaping the landscape of pharmaceutical analysis.
8.1 Green Chromatography
Environmental concerns and regulatory pressures have accelerated the adoption of green chromatography, which focuses on reducing the environmental impact of chromatographic analyses. This approach involves the use of eco-friendly solvents, such as water, ethanol, and supercritical CO?, instead of traditional organic solvents like acetonitrile and methanol, which are toxic and generate hazardous waste. Methods designed to minimize solvent consumption and waste generation contribute significantly to sustainability goals (Bhardwaj et al., 2020)[13]. The integration of supercritical fluid chromatography (SFC), which employs supercritical CO? as a mobile phase, exemplifies green chromatography by offering rapid separation with minimal solvent use and reduced energy consumption.
8.2 Microfluidic Liquid Chromatography Systems
The advent of microfluidic LC systems represents a major breakthrough in chromatographic technology by enabling the miniaturization of separation platforms. These lab-on-a-chip devices integrate multiple analytical processes on a single microchip, dramatically reducing sample volume, solvent use, and analysis time. Microfluidic systems offer high throughput, portability, and automation, making them highly suitable for point-of-care testing, rapid screening, and field applications. Their ability to perform on-chip separations allows for precise control over chromatographic conditions and enables complex sample analyses with minimal operator intervention.
8.3 Artificial Intelligence in Chromatography
Artificial intelligence (AI) and machine learning algorithms are increasingly applied to optimize chromatographic methods, data processing, and instrument control. AI-driven software can analyze vast datasets to predict optimal separation conditions, troubleshoot method development, and enhance peak identification. By automating routine tasks and improving decision-making accuracy, AI reduces method development time and improves reproducibility. This integration of AI is particularly impactful in high-throughput pharmaceutical environments where rapid method adaptation and robust data analysis are crucial.
8.4 Advances in Column Technology
The core of chromatographic separation lies in the column, and advances in column technology have greatly enhanced performance. Novel designs such as monolithic columns, core-shell particles, and nano-columns offer improved efficiency, resolution, and speed. Monolithic columns, composed of a continuous porous structure, provide high permeability and low backpressure, enabling fast separations with minimal solvent consumption. Core-shell particles combine a solid core with a porous shell, optimizing mass transfer and peak shape, thereby enhancing resolution and sensitivity. Nano-columns reduce particle size and column diameter to sub-micron levels, significantly increasing surface area and separation efficiency while lowering solvent usage. These innovations collectively contribute to higher throughput and greener analytical workflows (Bhardwaj et al., 2020)[14].
8.5 Future Perspectives
Looking ahead, the integration of multi-dimensional chromatography, hyphenated techniques such as LC-MS/MS and GC-MS/MS, and continued miniaturization will further enhance analytical capabilities. The convergence of chromatography with omics technologies (proteomics, metabolomics) and real-time monitoring systems is expected to revolutionize pharmaceutical analysis, enabling more comprehensive characterization of complex samples. Sustainability, automation, and AI will continue to be key drivers in the development of next-generation chromatographic platforms
Table: Recent Advances and Future Trends in Chromatography
|
Trend/Advance |
Description |
Benefits |
References |
|
Green Chromatography |
Use of eco-friendly solvents and supercritical CO? |
Reduced toxic waste, lower environmental impact, sustainability |
Bhardwaj et al., 2020 [13] |
|
Microfluidic LC Systems |
Lab-on-a-chip platforms for on-chip separations |
Reduced sample/solvent volume, portability, rapid analysis |
Bhardwaj et al., 2020 [13] |
|
AI in Chromatography |
Machine learning for method optimization and data analysis |
Faster method development, improved reproducibility |
Emerging technology |
|
Column Technology |
Innovations like monolithic, core-shell, and nano-columns |
Higher efficiency, resolution, lower solvent consumption |
Bhardwaj et al., 2020 [14] |
|
Future Perspectives |
Multi-dimensional chromatography, hyphenated techniques, integration with omics |
Enhanced analytical power, comprehensive sample characterization |
— |
CONCLUSION
Advanced chromatographic techniques are foundational to the modern pharmaceutical industry, providing the analytical precision, sensitivity, and reproducibility required to meet the rigorous demands of drug discovery, development, manufacturing, and regulatory compliance. As pharmaceutical products become increasingly complex—ranging from small molecules to biologics, biosimilars, and personalized medicines—the analytical methods used to evaluate them must evolve to offer higher resolution, faster throughput, and improved quantification accuracy. Techniques such as High-Performance Liquid Chromatography (HPLC), Ultra-High Performance Liquid Chromatography (UHPLC), Gas Chromatography (GC), and Supercritical Fluid Chromatography (SFC) have emerged as essential tools in this regard, each offering unique capabilities to address the wide spectrum of pharmaceutical analytical needs.One of the most important applications of these advanced techniques lies in method development and validation, which are critical steps in ensuring that analytical procedures are fit for their intended purpose. Method development involves optimizing chromatographic conditions such as the choice of stationary and mobile phases, flow rate, column temperature, gradient program, and detector settings. The goal is to achieve efficient separation of analytes, including the active pharmaceutical ingredient (API), excipients, impurities, degradation products, and residual solvents. Techniques such as UHPLC allow for reduced analysis time and increased resolution due to smaller particle sizes and higher operating pressures, making them particularly suited for high-throughput environments. Once an analytical method has been developed, it must undergo a rigorous validation process in accordance with international regulatory guidelines, including those from the International Council for Harmonisation (ICH Q2(R1)), the United States Pharmacopeia (USP), and the European Medicines Agency (EMA). Validation parameters such as specificity, linearity, range, accuracy, precision, limit of detection (LOD), limit of quantification (LOQ), robustness, and system suitability must be thoroughly evaluated. Advanced chromatographic techniques are particularly well-suited to meet these criteria due to their enhanced sensitivity and resolution capabilities, allowing for the accurate quantification of even trace-level analytes. In addition to their role in method development and validation, advanced chromatographic techniques are indispensable in the regulatory submission process. Data generated through validated chromatographic methods form the backbone of Chemistry, Manufacturing, and Controls (CMC) documentation submitted to regulatory authorities. These data support a wide range of regulatory requirements, including stability studies, batch release testing, bioequivalence studies, impurity profiling, dissolution testing, and pharmacokinetic evaluations. Without robust chromatographic analyses, it would be impossible to demonstrate the safety, efficacy, and quality of pharmaceutical products, particularly in the context of generic drug approvals and new drug applications (NDAs).Moreover, advanced chromatography plays a vital role in formulation development, enabling scientists to monitor the behavior of drugs under various physiological and environmental conditions. It is used to assess drug-excipient compatibility, track the formation of degradation products under stress testing, and evaluate the release profile of active ingredients in controlled-release formulations. In biologics and biosimilars, where molecular complexity poses a significant analytical challenge, chromatographic methods coupled with mass spectrometry (e.g., LC-MS/MS) are employed for structural elucidation, glycan profiling, and potency testing.As the pharmaceutical landscape continues to evolve, chromatography remains at the forefront of analytical science. Emerging innovations such as multidimensional chromatography (e.g., 2D-LC), hyphenated techniques (e.g., LC-MS, GC-MS), and green chromatography approaches using environmentally friendly solvents are pushing the boundaries of what can be achieved analytically. Additionally, advances in automation, artificial intelligence (AI), and machine learning are being integrated into chromatographic data analysis, enabling more efficient interpretation of complex datasets and reducing the likelihood of human error.In conclusion, advanced chromatographic techniques are not merely tools of separation—they are enablers of pharmaceutical innovation, quality control, and regulatory assurance. Their unmatched sensitivity, precision, and adaptability make them indispensable throughout the drug development lifecycle. As technological advancements continue to refine these methods, chromatography will remain a cornerstone of pharmaceutical science, driving improvements in drug quality, patient safety, and therapeutic efficacy well into the future.
REFERENCES










Rahul Chaudhari, Shweta Suryawanshi, Sneha Chitkalwar, Janhavi Patil, Ujwala Gawali, Sanket Biradar, Sarthak Sanap, Payal Jengthe, Pooja Hole, Harshada Deore, Advanced Chromatographic Techniques in Pharmaceutical Analysis, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 299-319. https://doi.org/10.5281/zenodo.15788069
 10.5281/zenodo.15788069
10.5281/zenodo.15788069