
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Faculty of Pharmacy, Bharath Institute of Higher Education and Research, Selaiyur, Chennai – 600073, Tamil Nadu, India.
Molecular docking is a pivotal computational approach in modern drug discovery, enabling the prediction of interactions between ligands and target proteins. However, the accuracy of docking outcomes depends on the quality of the input structures, necessitating a robust workflow that integrates homology modeling, protein preparation, and ligand preparation. This review aims to explore the current strategies in homology modeling, protein optimization, and ligand preparation to enhance molecular docking accuracy. It highlights key tools, methodologies, and recent advancements in in-silico modeling. Homology modeling is employed to construct reliable 3D structures of target proteins when experimental structures are unavailable. Techniques such as Swiss-Model and Modeller facilitate sequence alignment, model building, and structural validation. Protein preparation ensures the correction of missing residues, optimal protonation states, and energy minimization using tools like the Protein Preparation Wizard and Chimera. Ligand preparation involves optimizing chemical structures by generating 3D conformations, assigning charges, and performing energy minimization through Lig Prep and Open Babel. The integration of these preparatory steps significantly improves docking accuracy, leading to more precise predictions of binding affinity and molecular interactions. Recent advancements, such as machine learning-based scoring functions, flexible docking algorithms, and AI-driven ligand screening, further refine the docking process. A well-structured in-silico workflow for molecular docking enhances rational drug design by improving structure accuracy, reducing errors, and accelerating the drug discovery pipeline. Future developments in AI, quantum mechanics, and high-performance computing will further revolutionize the field, paving the way for more efficient and precise therapeutic discoveries.
Molecular docking is a cornerstone technique in computational biology and drug discovery, allowing the prediction of how small molecules, such as drugs, bind to specific biological targets, typically proteins. By understanding these interactions, researchers can identify and design potential therapeutic agents with high specificity and efficacy. [1]The process of molecular docking simulates the binding between a ligand and a target protein by evaluating the strength and nature of their interaction. Still, the accuracy and reliability of these predictions are entirely dependent on the quality of the input structures, i.e., the protein and the ligand. In this regard, a comprehensive workflow of homology modeling, preparation of the protein, and preparation of the ligand is necessary to ensure meaningful and accurate docking results. Homology modeling is a starting point in the absence of an experimentally determined 3D structure of a target protein by X-ray crystallography or NMR spectroscopy. This computational approach predicts the tertiary structure of the protein based on the structural knowledge of a homologous protein with known 3D structure, using which the conserved regions and structural motifs in the target protein's sequence are aligned with those in the template sequence. [2]Tools such as SWISS-MODEL, MODELLER, and Phyre2 are widely used in building homology models. These tools automate the model generation process but allow enough flexibility for manual intervention so that accuracy is improved after model generation. After generating the structures, stringent quality assessment is done using PROCHECK for Ramachandran plots, VERIFY3D for 3D structure compatibility, and QMEAN scores to ensure structural correctness and reliability. This validated model becomes the basis for any subsequent docking studies downstream. When a good quality 3D structure of the protein is in hand, protein preparation ensures the optimization of it to the docking study. [3] Generally, protein structures that were retrieved either by homology modeling or from experiments come with imperfections, which have missing residues, atoms, and sometimes loops, that are addressed. Lastly, solvent water molecules, ions, and ligands within the structure should also be assessed to determine relevance to the binding site. Preparation includes adding missing atoms, modeling missing loops and proper protonation states assignment of amino acid residues at physiological conditions. Energy minimization is carried out for removal of steric clashes and optimization of the protein's structure geometry. Specialized tools such as Schrödinger's Protein Preparation Wizard, Chimera, and AutoDock have been developed to automate this process. [4]The outcome of this step is a protein structure that is geometrically and energetically appropriate for docking. In parallel to protein preparation, ligand preparation is carried out to ensure the chemical correctness and biological appropriateness of the small molecules that are going to be docked. Ligands usually come from databases such as PubChem or ChemSpider or are drawn manually using tools like ChemDraw. Raw ligand structures have to undergo extensive processing in order to be able to represent their bioactive states under physiological conditions. These include generation of 3D conformations, partial charge assignment, tautomer and stereoisomer enumeration, and energy minimization to stabilize the structure. [5]Ligand preparation tools such as LigPrep, OpenBabel, and RDKit help automate these processes while allowing for customization according to specific requirements. A well-prepared ligand enhances the reliability of docking simulations by ensuring it is in its active conformation during binding. The integration of these preparatory steps—homology modeling for protein structure prediction, protein preparation for optimization, and ligand preparation for chemical accuracy—lays the groundwork for precise and meaningful molecular docking studies. Docking simulations involve the placement of the ligand in the binding site of the protein, generation of several binding poses, and ranking of the poses by scoring functions that estimate the binding affinity. [6] Visualization and analysis of the docking results give important information about the binding interactions, including hydrogen bonding, hydrophobic contacts, and van der Waals forces. Such information is very useful in identifying lead compounds, optimizing their properties, and designing novel therapeutics. The comprehensive approach of homology modeling, protein preparation, ligand preparation, and molecular docking accelerates the drug discovery process not only by accelerating it but also by cost and time consumption associated with experimental screening. [7]By developing advanced computational tools and methodologies, a researcher can actually explore any amount of the chemical space to predict accurately the protein-ligand interaction or prioritize those candidates for more experimental validations. This workflow is one of the powerful strategies in rational drug design that is being used in developing new treatments for diseases.
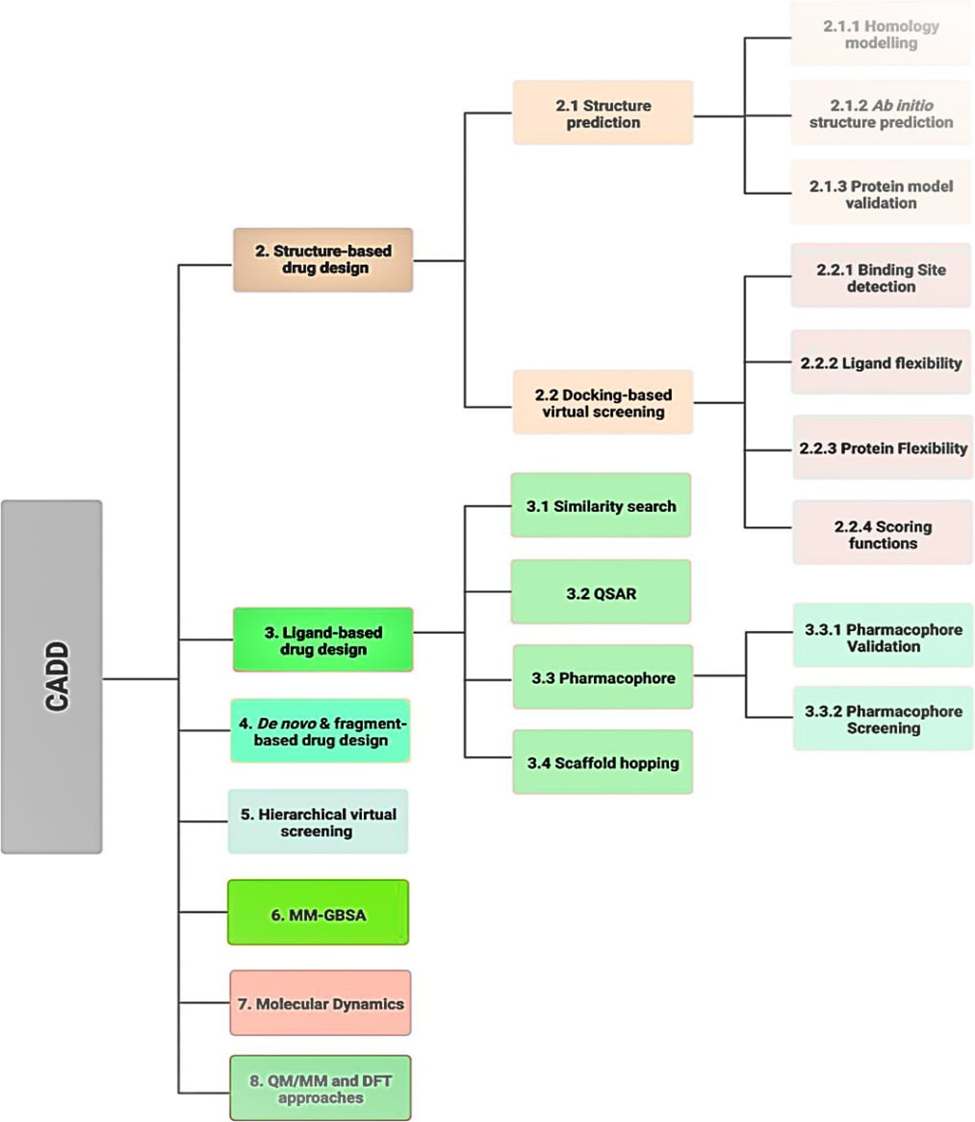
Figure 1: Various in silico techniques used in the drug design and discovery process discussed in this review (Abbreviations: CADD: computer-aided drug design; DFT: density functional theory; MM: molecular mechanical; MM-GBSA: molecular mechanics with generalized born and surface area; QM: quantum mechanical; QSAR: quantitative structure activity relationship).
Recent Advances in In-Silico Molecular Docking
Molecular docking has significantly improved over the past years due to the advancements in computational power, algorithms, machine learning, and integration with experimental techniques. [8]These developments have enhanced the precision, efficiency, and scope of docking studies, making them invaluable tools in drug discovery and structural biology. Below is a detailed view of recent advances in in-silico molecular docking: Thanks to developments in artificial intelligence, algorithm development, computer capacity, and integration with experimental methods, molecular docking has made significant strides in recent years.[9] These advancements have improved docking studies' precision, speed, and adaptability, making them essential to structural biology and contemporary drug development. Scoring functions, which are crucial for estimating ligand-receptor affinities and grading binding poses, are one of the main areas that need to be improved. Machine learning-based approaches such as RF-Score and DeepDock, which use massive datasets of experimental binding affinities to train predictive algorithms, are increasingly a part of recent scoring strategies. [10]Furthermore, physics-based models now take into account intricate interactions like solvation, entropy, and induced fit effects, and consensus scoring—which combines several scoring techniques—helps lessen individual bias and increase dependability. Protein flexibility has also changed how it is treated. The predictive power of earlier docking techniques was limited by their assumption of a rigid protein structure. More realistic simulations are now possible thanks to methods like flexible receptor docking algorithms like RosettaDock and AutoDockFR, molecular dynamics (MD)-integrated docking, and Induced Fit Docking (e.g., Schrödinger’s IFD). [11] Biomolecular flexibility modeling has been further enhanced by ensemble docking, which entails docking ligands into numerous protein conformations derived from MD simulations, NMR ensembles, or cryo-EM data. Computational advancements have also helped in ligand screening and preparation. More effective chemical space exploration is made possible by fragment-based docking, in which smaller ligand fragments are docked and then assembled. AI-based filters are currently included into improved virtual screening techniques to screen large compound libraries, such as the ZINC and ChEMBL databases, with high throughput. De novo drug design technologies, such as AutoGrow and DeepDrug, use generative algorithms to create new ligands that are tailored for certain protein targets.[10] Throughout the docking operation, artificial intelligence is essential, especially in position prediction, where deep learning models perform better than conventional techniques by more accurately capturing intricate molecular interactions. To iteratively improve docking predictions and suggest new ligand structures, methods like active learning and generative models—such GANs and variational autoencoders—are being employed.Due to these advancements, docking is now possible for formerly challenging targets including RNA molecules, big, flexible ligands like macrocycles, and intrinsically disordered proteins (IDPs). The identification of non-orthosteric modulators is aided by sophisticated algorithms that currently anticipate binding at allosteric sites. Moreover, the incorporation of docking with experimental methods has grown in popularity and strength. [11] Computational predictions are validated and improved by hybrid approaches that include data from cryo-EM, NMR, or X-ray crystallography into docking operations. To identify precise binding locations and enhance ligand selection, structure-based pharmacophore modeling and cryo-EM-guided docking are employed. Large-scale docking is becoming more feasible and effective because to the growth of cloud computing and high-performance computing. Cloud services like AWS and Google Cloud facilitate high-throughput docking processes, while distributed computing platforms like Rosetta@home and Folding@home enable the utilization of worldwide computational resources. Faster processing is made possible by GPU-accelerated software, such as AutoDock-GPU, and quantum computing, while still in its infancy, holds promise for resolving intricate quantum mechanical issues in ligand-receptor interactions. [12]Lastly, multi-target and multi-objective docking techniques have gained more attention.These methods are particularly helpful in polypharmacology, the study of ligands that act on several disease-relevant targets. Additionally, pharmacokinetic and toxicity profiles (ADMET) are now frequently included in optimization along with binding affinity, which aids researchers in finding more clinically viable drug candidates. [13] Recent advancements in docking software, including AutoDock-GPU, DeepDock, and hybrid tools like Schrödinger Glide and GOLD, have improved molecular docking's performance, accuracy, and usability, solidifying its position as a fundamental component of computational drug development.
Homology Modeling For Molecular Docking
Homology modeling, also known as comparative modeling, is a computer-based prediction of the three-dimensional (3D) structure of a protein for which no experimental structure has been determined. This relies on the fact that proteins whose sequences are similar have very similar structures.[14] It's an essential tool for molecular docking since it offers a structural framework that will aid in understanding protein-ligand interaction. The detailed discussion of principles, steps, tools, challenges, and applications of homology modeling in molecular docking are described below. When no experimentally determined structure is available, homology modeling—also known as comparative modeling—is a computational method used to predict a protein's three-dimensional (3D) structure. [15] The foundation of this approach is the idea that proteins with comparable amino acid sequences typically have structural conformations. Because it offers a structural foundation for comprehending protein-ligand interactions, this method is essential to molecular docking. [16]A few fundamental presumptions form the basis of homology modeling. First of all, proteins with somewhat similar sequences typically take on folds that are comparable. Second, specific structural motifs frequently correlate with local sequence motifs. [17]Thirdly, essential protein residues, particularly those found in active regions, are typically conserved among homologous proteins. Under these presumptions, a target protein's unknown structure can be modeled by matching its sequence to a known structure (template) and building a model in accordance with that alignment. A number of crucial processes are involved in the homology modeling process. The first step is template selection, in which BLAST or HMMER are used to match the target protein sequence with structural databases such as the Protein Data Bank (PDB). Sequence identity (usually better than 30%), similarity, and structural resolution are among the factors used to select templates. Following the selection of an appropriate template, the target sequence is aligned with the template using alignment tools such as Clustal Omega or T-Coffee in order to map conserved and variable sections. [18] This process is known as sequence alignment. After alignment, the target's amino acids are substituted for the template's while maintaining the general backbone structure to create the model. Loop modeling methods or ab initio techniques are used to model regions like loops or termini that lack a corresponding template. Optimization, frequently using energy reduction techniques, is necessary to fix structural errors such as torsional angle deviations and steric conflicts once the basic model has been built. The precision of flexible regions is improved by using loop modeling tools like as MODELLER or ROSETTA. The final structure is subsequently validated using programs such as QMEAN, Verify3D, and PROCHECK (Ramachandran plot analysis) to evaluate the model's structural and geometric integrity. [19]To increase reliability, errors found during validation—particularly in areas with inadequate modeling—are further improved. Homology modeling is supported by a number of software tools, such as Phyre2 and RaptorX, which are especially useful for detecting distant homologies or modeling proteins with low sequence similarity; MODELLER, which is renowned for its adaptability in structure prediction; I-TASSER, which combines homology and ab initio modeling; and SWISS-MODEL, which provides a fully automated pipeline.Despite its benefits, homology modeling has significant drawbacks. [20]When the target shares low sequence identity (<30%) with known structures, choosing suitable templates becomes challenging and may lead to inaccurate models. Inaccurate structural predictions could result from alignment mistakes made during the sequence-template comparison. Furthermore, the lack of template information makes it challenging to adequately describe changeable sections like loops or insertions. [21] Without experimental direction, side-chain conformation prediction is still difficult, particularly in functionally important active areas. Furthermore, it is intrinsically challenging to validate the accuracy of the modeled structure in the absence of experimentally determined structures for comparison. Homology modeling has greatly improved in recent years. For large protein complexes, improved structural predictions are obtained by combining homology modeling with data from cryo-electron microscopy (cryo-EM). [22] Tools powered by artificial intelligence (AI), such as RoseTTAFold and AlphaFold, have transformed the discipline by producing incredibly accurate models, even for proteins without close homologs. In order to improve accuracy, particularly for flexible or poorly conserved areas, hybrid modeling systems that combine homology modeling with ab initio techniques or molecular dynamics simulations are now commonly employed. [23]Additionally, restrictions in template-free regions have been overcome by advancements in loop modeling and de novo prediction, leading to more comprehensive and trustworthy homology models.
Protein Preparation for Molecular Docking:
Protein preparation is a crucial step in molecular docking, where the target protein structure will be optimized to be well-suited for docking simulation. [24]Raw protein structures can be obtained from various techniques such as X-ray crystallography, NMR spectroscopy, and sometimes computational modeling. [25]Invariably, these raw structures contain imperfections like missing residues, ambiguous protonation states, and even crystallization artifacts. Protein preparation ensures that the protein is present in its biologically meaningful state, with an energetic conformation that will most accurately represent its binding site. Below is a detailed breakdown of the process of protein preparation, including its importance, workflow, tools, challenges, and recent advancements. In molecular docking, protein preparation is an essential step that forms the basis for precise ligand-binding predictions. [26] Prior to docking simulations, it is necessary to fix structural flaws in raw protein structures, which are usually obtained by computational approaches like homology modeling or experimental methods like X-ray crystallography or NMR spectroscopy. Inaccurate bond geometries, unclear protonation or tautomeric states, missing atoms or residues, and extraneous elements like ions, co-crystallized ligands, or crystallographic water molecules that are unrelated to the intended docking study are some examples of these flaws. [27]In order to increase the accuracy of predicted binding interactions, protein preparation aims to guarantee that the target protein has a physiologically realistic shape. The first step in the procedure is to either generate the proper protein structure computationally or retrieve it from sources such as the Protein Data Bank. [28] The right biological assembly—whether a monomer, dimer, or higher-order complex—is next confirmed by cleaning the structure to eliminate non-biological components, as this influences the precision of docking results. The structural continuity is then restored by modeling in any missing heavy atoms, hydrogens, or whole residues, especially in loop regions, using specialized tools. Using pKa prediction methods like PROPKA or H++, precise protonation and charge assignment are carried out for ionizable residues like histidine, lysine, glutamate, and aspartate based on physiological pH (about 7.4). [29] Histidine receives particular attention because of its many protonation and tautomeric forms, which might affect interactions with ligands. Energy minimization is used to ease steric conflicts, adjust bond angles, and maximize the protein's shape after the protonation states have been determined. In order to maintain the overall protein structure while permitting local modifications, this is typically accomplished using molecular mechanics force fields like AMBER, CHARMM, or OPLS with constrained minimization. [30]Next, information from conserved residue studies, mutagenesis experiments, co-crystallized ligands, or computational site-prediction tools such as SiteMap and CASTp are used to define the binding site. Each water molecule is assessed separately; some are eliminated if they don't aid in ligand binding, while others that mediate crucial hydrogen bonds close to the active site might be kept. The entire protein structure, including cofactors or metal ions that need to be handled carefully to guarantee compliance with the docking technique, is given force field parameters. It is crucial to validate the generated protein structure, usually with the use of programs like Verify3D to check for structural compatibility or PROCHECK, which offers a Ramachandran plot to gauge stereochemical quality. The entire protein preparation process is streamlined by a number of technologies and software platforms. [31] One of the most complete is Schrödinger's Protein Preparation Wizard, which permits hydrogen addition, structure optimization, and constrained minimization.[32] While AutoDockTools (ADT) is designed for AutoDock and Auto Dock Vina preparation, Chimera and ChimeraX include visualization and manual editing features. MOE and Discovery Studio offer sophisticated tools for structure cleaning, site identification, and reduction, while PDBFixer offers fast fixes for missing atoms and residues. [33]The accurate handling of incomplete structural data, ambiguity in protonation states, the difficult choice of keeping or removing water molecules, the correct parameterization of metal ions, and identifying the correct binding site in proteins with multiple potential cavities are just a few of the difficulties that arise during protein preparation even with these advanced tools. [34] Recent developments, however, are resolving these problems by using artificial intelligence, which is better able to forecast protonation states and tautomeric forms, and by improving the quality of models by refining cryo-electron microscopy (cryo-EM) data. High-throughput preparation pipelines have been made possible by automation and cloud-based technologies, and molecular dynamics simulations are being used more and more to prepare proteins in dynamic conformations that capture the flexibility of side chains or loops. Furthermore, post-translational modifications (PTMs) like phosphorylation, glycosylation, or methylation are starting to be supported by contemporary techniques.[35] These modifications are essential for realistic docking investigations involving biologically changed proteins. The success and interpretability of molecular docking investigations are directly impacted by the complex and dynamic process of protein synthesis.
Ligand Preparation for Molecular Docking
Ligand preparation is an important step in molecular docking that ensures ligands are in their most suitable chemical form for accurate docking and interaction analysis with the target protein.[36] Raw ligand structures, often drawn up from chemical databases or sketched manually, might be missing optimized geometries, protonation states, or defined stereochemistry, all of which can result in erroneous docking results. [37]Therefore, proper ligand preparation addresses these issues and thereby provides reliable and biologically meaningful docking predictions. Ligand preparation involves optimizing the 3D structure of ligands by assigning correct bond orders, stereochemistry, tautomeric forms, and protonation states. [39]The process also includes energy minimization to resolve structural strain and ensure compatibility with the docking software. Tools like Open Babel, Schrödinger’s LigPrep, and AutoDockTools facilitate these tasks. The quality and dependability of computer predictions about ligand–protein interactions are strongly impacted by ligand preparation, a fundamental and essential stage in molecular docking. [40] Raw ligand structures are usually obtained from chemical databases like PubChem, ZINC, or ChEMBL or manually sketched using drawing programs like Chem Draw or Marvin Sketch in drug discovery and molecular modeling workflows. However, these initial representations often lack the critical chemical and structural refinements required for successful docking. [41] Common problems that can cause major errors in the docking process and ultimately lead to false biological interpretations include wrong or undefined bond ordering, missing hydrogen atoms, inappropriate stereochemistry, and non-optimized 3D geometry. [42] In order to ensure that each ligand is in its most chemically and physiologically significant state prior to being put through docking simulations, ligand preparation helps to refine these structures. In the initial phase of ligand preparation, the ligand structure is cleaned and input. Depending on the docking software to be used, the raw ligand must be transformed into an appropriate file format, such as MOL2, SDF, or PDB, regardless of the source.[43] Accurate downstream processing is made possible by these formats' preservation of structural and chemical data. The ligand then goes through standardization, which includes resolving stereochemistry ambiguities, correcting valence mistakes, assessing atom connectivity, and assigning the proper bond ordering (e.g., single, double, aromatic). [44] Through this procedure, the molecule's chemical accuracy and desired configuration are guaranteed. Particularly, the correct assignment of stereochemistry is essential when dealing with chiral centers, as the biological activity of enantiomers can vary drastically.In order to automate many of these modifications and expedite the input standardization process, tools such as Open Babel, RDKit, and Schrödinger's LigPrep are frequently utilized in this stage. The assignment of protonation states is a crucial step after structural standardization. Depending on the pKa of their functional groups, many ligands exist in ionized forms at physiological pH (~7.4). The binding affinity and orientation of ligands at the protein's active site can be greatly impacted by the precise prediction and assignment of the protonation state for groups like carboxylic acids (-COOH), amines (-NH2), phenols, and imidazoles. Inaccurate electrostatic interactions or hydrogen bonding networks, which are essential for molecule recognition, could result from errors in this stage. [45]In order to automate many of these modifications and expedite the input standardization process, tools such as Open Babel, RDKit, and Schrödinger's LigPrep are frequently utilized in this stage. The assignment of protonation states is a crucial step after structural standardization. Depending on the pKa of their functional groups, many ligands exist in ionized forms at physiological pH (~7.4). The binding affinity and orientation of ligands at the protein's active site can be greatly impacted by the precise prediction and assignment of the protonation state for groups like carboxylic acids (-COOH), amines (-NH2), phenols, and imidazoles. Inaccurate electrostatic interactions or hydrogen bonding networks, which are essential for molecule recognition, could result from errors in this stage. Software programs like as Open Babel, ChemAxon's Marvin Sketch, and Schrödinger's Epik aid in predicting and applying the proper protonation states under biologically relevant circumstances. [46] The creation and choice of tautomeric forms are closely associated with this. Numerous bioactive substances, like keto-enol pairings, can exist in several tautomeric forms, each of which may have unique binding characteristics. Generating every biologically feasible tautomer and assessing them for docking, either by hand or with automated screening technologies, is crucial. The ligand's 2D structure is transformed into a 3D conformation after the proper protonation and tautomeric forms have been chosen. Since it establishes how the ligand fits into the target protein's binding pocket, this three-dimensional model is crucial for spatially correct docking.Using cheminformatics tools like RDKit, LigPrep, or Open Babel, which create plausible molecular structures based on known geometrical parameters and bonding patterns, initial 3D geometries can be produced. However, these structures need to be fixed before docking since they could have high internal strain, steric conflicts, or unfavorable torsions. Consequently, energy minimization utilizing molecular mechanics force fields like MMFF94, OPLS, or CHARMM is the next step. [47] A more stable and realistic conformation is produced by energy minimization, which modifies bond angles and torsions to reach a local energy minimum. By making sure the ligand is energetically and geometrically compatible with the molecular docking algorithm, this step increases the reliability of docking. The precise control of chirality and stereoisomerism is another crucial component of ligand synthesis. Because distinct stereoisomers might have radically different binding mechanisms and pharmacological characteristics, chiral centers need to be clearly described. It could be necessary to synthesize and dock each potential stereoisomer separately if the ligand has unknown chiral centers. The same is true of cis/trans isomerism in double bonds, which can affect a molecule's structure and binding capacity. [48] Furthermore, a large range of potential conformations are made feasible by the numerous rotatable bonds that flexible ligands frequently possess. A library of conformers is created using either stochastic techniques (like RDKit, OMEGA) or systematic techniques (like ConfGen) to allow for this flexibility. During docking, these conformers are utilized to investigate various binding poses and offer a sample of the molecule's potential spatial configurations. Converting the ligand structure into a file format that works with the docking software is the last stage of ligand preparation. For example, PDBQT files, which contain torsion tree data for flexible docking, are necessary for AutoDock and AutoDock Vina. Whereas DOCK uses MOL2 formats, Glide uses MAE (Maestro) files. Torsion flexibility, partial atomic charges, and atom kinds must be specified during this conversion in accordance with the docking engine's specifications. It is wise to use molecular visualization tools (such as PyMOL, Chimera, or Maestro) to visually and computationally validate the generated ligand after the structure is finalized. This will ensure that the structure is consistent, that it is oriented correctly, and that no artifacts were introduced during processing. To guarantee the accuracy of docking data, a number of crucial factors need to be taken into account throughout the ligand production process. According to rules like Lipinski's Rule of Five, which takes into account variables like molecular weight, lipophilicity (logP), and hydrogen bond donors and acceptors, the physicochemical characteristics of ligands should fall within acceptable limits of drug-likeness. [50] Ligands with characteristics outside of this range might not be well absorbed or bioavailable. Because erroneous states might result in docking poses that are physiologically inappropriate, tautomeric and protonation variability must be carefully regulated. When making ligands that interact with metalloproteins, more care must be taken. It can be difficult to simulate these ligands using classical force fields because they frequently contain metal-chelating groups that need precise modeling of metal coordination geometry and partial charges. The ligand preparation procedure has been greatly enhanced by recent developments. To improve the speed and precision of this phase, artificial intelligence and machine learning models are being created to forecast the most likely tautomeric, protonation, and conformational states of ligands. [51] High-throughput analysis of sizable compound libraries is now possible because to automated processes and pipelines that combine ligand production with virtual screening techniques, as those offered by Schrödinger's suite. Additionally, because molecular dynamics (MD) simulations help capture the dynamic behavior and flexibility of ligands in a biologically plausible context, they are increasingly being used in ligand production. Optimization techniques based on quantum mechanics (QM) are also becoming more popular because they can refine geometries and assign partial charges more accurately than traditional techniques. In structure-based drug design (SBDD), where accurate modeling of ligand-receptor interactions is crucial, these advancements are especially significant. [52] Similar to this, properly produced ligand fragments enable more accurate reconstruction of larger bioactive molecules and effective probing of binding sites in fragment-based drug design (FBDD). In the end, better docking accuracy, accurate binding affinity predictions, and more effective lead compound identification in computational drug development are all influenced by high-quality ligand production.
Biological And Ligand Databases for Molecular Docking
Databases play a pretty significant role in molecular docking studies, providing essential biological and chemical information including protein structures, ligand libraries, and bioactivity data. Detailed key databases used in molecular docking workflows are listed as follows. The foundation of molecular docking investigations is databases, which provide an organized and extensive store of chemical and biological data essential to each step of the docking process. These databases greatly improve the precision, repeatability, and reach of computational drug discovery efforts by facilitating the efficient retrieval, analysis, and application of data pertaining to target proteins, ligand libraries, bioactivity data, structural interactions, and metabolic pathways. When choosing appropriate macromolecular targets for docking, biological databases—especially those that store protein structures—are invaluable. [53] The *Protein Data Bank (PDB), which contains experimentally determined three-dimensional structures of proteins, nucleic acids, and macromolecular complexes acquired by nuclear magnetic resonance (NMR), cryo-electron microscopy (cryo-EM), and X-ray crystallography, is one of the most basic resources.In order to facilitate the precise construction of docking targets and grid definitions, PDB entries are enhanced with structural metadata, such as active site annotations, cofactors, and ligands co-crystallized with target proteins. Computational databases, however, bridge the gap for many proteins for which there are no crystal structures accessible. Developed with the groundbreaking AlphaFold AI technology, the AlphaFold Protein Structure Database offers highly accurate predicted protein structures that have proven useful in structure-based drug design, especially for orphan targets. [54] Similar to this, the Swiss Model Repository uses sequence similarity to established templates to anticipate the structure of proteins by providing homology-modeled structures for those that lack experimental evidence. The range of docking targets can be greatly increased by using these projected or modeled structures, particularly in the case of poorly described proteins, neglected diseases, or newly developing infectious diseases. Resources such as UniProt, a carefully curated and extensive database comprising protein sequences, domains, post-translational modifications, and functional annotations, provide sequence-level information and functional annotations in addition to structural databases. By providing information on protein function, subcellular localization, and pathway involvement, UniProt makes it possible to choose targets that are physiologically significant. **BindingDB is an important database that lists experimentally observed binding affinities, such as IC??, Kd, and Ki values, for protein-small molecule complexes, providing more insights into molecular interactions. In addition to helping with ligand selection, this database is essential for confirming docking results since it offers reference experimental data for comparing projected binding scores. [55]The exact localization of docking grids is supported by tools like CASTp (Computed Atlas of Surface Topography of proteins), which let researchers locate and analyze the shape and volume of active sites, pockets, and cavities on protein surfaces. PDBsum also provides annotated summaries of ligand-binding diagrams, hydrogen bonds, and hydrophobic contacts—all of which are examples of protein-ligand interactions seen in PDB structures. Designing docking studies that accurately depict biological interactions and interpreting docking results within a structural framework require this kind of knowledge.
Ligand databases, which provide the tiny compounds to be docked against the target proteins, are equally important to molecular docking.One of the biggest databases of chemical molecules is PubChem, which is hosted by the National Center for Biotechnology Information (NCBI). [56]It provides millions of 2D and 3D compound structures, together with information on their physicochemical characteristics and biological experiment results. PubChem is widely used to generate a variety of virtual libraries for screening and to get ligands with known biological activity. The Irwin and Shoichet labs at UCSF maintain the ZINC database, which provides carefully selected, ready-to-dock libraries of small compounds that are readily available in the market and formatted for instant use in docking tools. To correspond with different phases of virtual screening campaigns, it contains a variety of subsets, including lead-like, fragment-like, and drug-like libraries to correspond with various virtual screening campaign stages. ChEMBL, a manually maintained database of bioactive compounds with drug-like qualities that connects chemical structures to their pharmacological targets and related bioactivity measurements, is another popular database. By offering a comprehensive collection of small compounds with in-depth assay annotations, ChEMBL facilitates lead optimization, docking validation, and structure-activity relationship (SAR) research. [57] DrugBank is a vital resource for research on authorized medications and their repurposing for novel therapeutic targets. By combining comprehensive pharmacological, chemical, and molecular target data on both FDA-approved and experimental medications, it makes it possible to screen for repositioning candidates in silico, evaluate druggability, and anticipate off-target interactions. Molecular docking also makes extensive use of natural compounds, which are renowned for their biological potency and structural diversity. Docking research centered on nature-derived medicines are made possible by specialized databases like NPASS (Natural Product Activity and Species Source Database), which gather natural compounds from different species along with their known bioactivities. A comprehensive list of natural chemicals and their derivatives that can be utilized for virtual screening is also provided by SuperNatural II. [58] The Traditional Chinese Medicine Database (TCMD) offers natural compounds derived from traditional Chinese medicinal plants, herbs, and formulations for traditional medicine-based docking, particularly in ethnopharmacological research. This facilitates the integration of traditional knowledge with contemporary computational techniques. These databases of natural products aid in the discovery of new scaffolds with possible therapeutic applications and make it easier to explore the chemical space outside of synthetic libraries. Databases of tiny molecule fragments are the foundation of fragment-based docking techniques, which are being utilized more and more in early-stage drug development. Libraries of fragment-like and lead-like compounds that are commercially available for fragment-based screening and hit expansion are provided by eMolecules and Enamine REAL. [59] After early docking hits, these fragments can be used as building blocks for de novo drug discovery or to develop into more complex, higher-affinity ligands. End-to-end workflows from ligand identification to docking validation are made possible by certain databases, such BindingDB, which fulfill two functions by combining protein and ligand data with related experimental binding data. The PDB-affiliated RCSB Ligand Expo lists ligands co-crystallized in PDB entries, providing useful information on bioactive conformations and binding modalities that support docking pose assessment and scoring. These databases are used in a variety of molecular docking workflow contexts. Biological databases aid in the prioritization and selection of pertinent proteins during target identification by considering factors like as sequencing, structure, and functional significance. Chemical and natural product databases offer a variety of libraries that support high-throughput virtual screening campaigns in ligand screening. [60]Co-crystallized structures and experimentally determined binding affinities from integrated databases serve as validation, confirming the accuracy of anticipated docking postures and binding scores. Databases like Drug Bank, which associate current medications with a variety of targets, support drug repurposing initiatives by facilitating economical repositioning tactics. Last but not least, natural product docking finds new bioactive compounds with traditional origins by utilizing pharmacological and ethnobotanical data from NPASS, TCMD, and Supernatural II. In conclusion, these databases work together to create an essential infrastructure that makes molecular docking analyses effective, repeatable, and informative. This greatly speeds up drug development and aids in the logical creation of novel therapeutic agents.
Different Software Tools Applied for Molecular Docking
Molecular docking relies on computational tools that estimate how small molecules (ligands) interact with a macromolecule, often a protein. [61]The disparity in their algorithms, features, and simplicity makes these tools suited to solve specific needs during docking studies. Some of the most commonly used software for molecular docking are shown below. A key method in structure-based drug design is molecular docking, which allows scientists to predict a ligand's preferred orientation when it is attached to a target macromolecule—typically a protein—in order to clarify the type and intensity of the interaction. In order to meet a variety of research goals, this procedure heavily relies on computational tools, which vary in terms of their algorithms, degrees of flexibility, scoring accuracy, user interfaces, and processing demands. Open-source docking software is very common in scholarly research because of its versatility and ease of use. [62]Flexible docking is made possible by tools such as AutoDock, which use a Lamarckian Genetic Algorithm (LGA) to enable both the ligand and the receptor to investigate conformational changes during interaction. By performing flexible docking using a Lamarckian Genetic Algorithm (LGA), tools such as AutoDock enable both the ligand and the receptor to investigate conformational changes during interaction. AutoDock-GPU uses graphics processing units to greatly speed up the docking procedure, while AutoDock Vina, its variation, delivers quicker performance through multithreading and a streamlined scoring algorithm. Similarly, one of the first docking programs to be developed, DOCK, currently allows both rigid and flexible docking and focuses on receptor-ligand matching using cavity-based techniques. [63] While rDock is praised for its adaptable procedures appropriate for virtual screening and lead tuning, OpenEye FRED offers comprehensive conformational sampling for stiff docking, guaranteeing quick and precise pose prediction.By combining AutoDock and Vina into an intuitive interface, GUI-based technologies such as PyRx streamline the docking process, making it effective for high-throughput screening and accessible to novices. However, because of their sophisticated algorithms, excellent user assistance, and wealth of features, commercial software platforms are widely employed in the pharmaceutical industry and in prestigious university research. For instance, Schrödinger Glide is perfect for large-scale screening and comprehensive binding affinity predictions since it provides customizable ligand docking and high-precision docking based on empirical scoring. A complete suite for molecular modeling, visualization, and simulation is offered by MOE (Molecular Operating Environment), which also supports numerous scoring approaches and flexible docking.[64] The CDOCKER algorithm, which is part of Discovery Studio, powered by BIOVIA, improves the accuracy of interaction prediction by enabling precise docking through molecular dynamics-based refinement. With a focus on electrostatic and form complementarity between ligands and receptors, Cresset Spark and Blaze present a field-based method that is especially helpful for scaffold hopping and virtual screening. In addition to general-purpose docking tools, specialized and hybrid software solve particular problems in the industry. For flexible docking, GOLD (Genetic Optimization for Ligand Docking) uses genetic algorithms, which perform especially well in situations with intricate and limited binding pockets. [65] FlexX is very good at screening big chemical libraries because it uses fragment-based docking, which builds ligands gradually inside the active site.In the context of macromolecular interactions, HADDOCK (High Ambiguity Driven DOCKing) is unique in that it improves prediction accuracy for protein-protein and protein-DNA complexes by integrating experimental data, such as NMR constraints, into docking techniques. Furthermore, Rosetta Flex Pep Dock* is an expert in peptide-protein docking, which enables side-chain and backbone flexibility to precisely simulate dynamic interactions. Computational docking has become even more accessible with the rise of web-based tools like as *DockThor, **Mcule, **GNINA, and *SwissDock. Researchers can use these platforms to complete docking activities without requiring sophisticated computer hardware. While DockThor offers flexible and blind docking modes via an easy-to-use online interface, SwissDock leverages the CHARMM force field and is coupled with SwissSidechain for ligand customisation. Molecule is a simplified online platform that integrates docking, virtual screening, and ligand synthesis. By integrating deep learning models into its docking pipeline, GNINA distinguishes itself and greatly improves posture prediction and scoring, particularly in high-throughput scenarios. Additionally, researchers are increasingly using molecular dynamics-integrated docking tools like CHARMM-GUI and AMBER to create better realism in docking simulations. [66] These platforms capture conformational flexibility and solvent effects that static docking approaches could miss by simulating the dynamic behavior of ligands and binding sites throughout time. While CHARMM-GUI makes it easier to set up complex systems for MD simulations, including membrane-bound proteins, and supports improved sampling techniques for better conformational exploration, AMBER integrates energy minimization, molecular mechanics, and MD simulations to provide a deeper understanding of ligand-receptor interactions. The type of molecular target—whether it involves small molecules, proteins, or nucleic acid complexes—as well as the required degree of flexibility, scoring function accuracy, usability (GUI vs. command-line interface), availability of computational resources (CPU vs. GPU), and financial constraints must all be carefully taken into account when choosing the right docking software. [67] GPU-accelerated or ML-enhanced platforms, for instance, may be advantageous for high-throughput virtual screening campaigns, but experimental data-driven or MD-integrated tools may be necessary for in-depth mechanistic investigations. Researchers can obtain precise predictions of binding modes and affinities by matching the tool's capabilities with the scientific goals of a docking study. This will speed up the drug discovery process and improve our comprehension of biomolecular interactions.
Molecular Docking: Detailed Steps
Molecular docking is a prediction of the preferred orientation of a ligand bound to a target macromolecule, usually a protein.[68] The process imitates real-life molecular interactions and thus can offer insights into binding affinity, stability, and molecular recognition. Below is a detailed explanation of the key steps in molecular docking: A potent computer method called molecular docking is used to forecast the preferred orientation and mode of binding of a ligand—a tiny molecule—when it interacts with a biological macromolecule, usually a protein. By simulating actual molecule interactions, this procedure offers important insights into stability, binding affinity, and the molecular recognition mechanisms underlying biological activities. The first phase in the multi-step docking workflow is target preparation, in which the biological receptor—usually an enzyme, receptor, or protein—is chosen from structural databases like Swiss Model, AlphaFold, or the Protein Data Bank (PDB). Accurate and significant docking results depend on the target being properly prepared. [69]This entails clearing the structure of ions that could obstruct the docking process, non-essential heteroatoms, and water molecules (unless they are needed for ligand binding). To replicate appropriate hydrogen bonding, missing hydrogens—especially polar ones—must be added. After that, any steric conflicts are fixed and bond angles and geometries are improved by optimizing the structure using molecular mechanics force fields like CHARMM or AMBER. Another important step is determining the binding site, which is usually accomplished by hand inspection of co-crystallized ligands or by employing structural databases and analytical tools such as CASTp or PDBsum. Blind docking can be used to examine the receptor's whole surface if the binding site is unknown. To make sure the ligand is in the proper conformational, tautomeric, and protonation states, ligand preparation is then carried out. Ligands may be obtained from well-known inhibitors, natural substances, or large virtual libraries such as ZINC, PubChem, and ChEMBL. 2D structures must first be transformed into 3D conformations, and then energy and geometry must be optimized using computational tools like AMBER and MMFF94 for refinement based on molecular mechanics or Gaussian for quantum mechanical computations. Additionally, the ligand needs to be modified to represent its proper protonation state at physiological pH (~7.4), which can be done with programs like MarvinSketch or OpenBabel. All of the molecule's physiologically active forms should be taken into account by taking into account pertinent tautomers and stereoisomers.The process of grid formation involves defining the spatial area in which the ligand will be docked by creating a grid box around the active site. [70] This grid must contain all essential residues involved in binding and is defined using the docking program. The distance between grid points, which is typically set to approximately 0.375 Å in programs like AutoDock to balance accuracy and computing performance, determines the grid's resolution. This grid creates energy maps for various atoms (such as carbon, oxygen, nitrogen, etc.) to direct the ligand during docking. These maps are based on potential interaction energies that are determined using pertinent force fields. The software method samples several ligand orientations and conformations with respect to the binding pocket in order to investigate several binding modalities throughout the *docking process. To investigate the ligand's conformational space, algorithms including Molecular Dynamics (Glide), Monte Carlo simulations (DOCK), and Genetic Algorithms (AutoDock) are used. Scoring functions are used to assess binding affinity based on electrostatic interactions, van der Waals forces, desolvation penalties, and total binding free energy for each sampled pose. This procedure is further improved by flexible docking techniques, which provide movement to the ligand and, in certain situations, the receptor, increasing the simulation's resemblance to actual biological settings. [70]The most promising ligand-receptor complexes are found and examined through post-docking analysis once docking is finished. This entails choosing the binding postures that have the best scores and employing molecular visualization software such as Discovery Studio, UCSF Chimera, or Py MOL to visualize these poses. The most promising ligand-receptor complexes are found and examined through post-docking analysis once docking is finished. This entails choosing the binding postures that have the best scores and employing molecular visualization software such as Discovery Studio, UCSF Chimera, or PyMOL to visualize these poses. To learn how the ligand stabilizes inside the binding pocket, important interactions such salt bridges, hydrophobic contacts, π–π stacking, and hydrogen bonds are closely examined. In order to highlight regular binding patterns and facilitate interpretation, clustering techniques are frequently employed to group comparable binding postures. docking outcome validation is required to guarantee the accuracy of docking predictions. In order to verify the accuracy of the software and parameters utilized, this entails redocking known co-crystallized ligands into their original binding locations and comparing the predicted poses to the ones found experimentally. [71] The specificity and robustness of the docking process can be tested using negative controls, such as inert compounds or structural decoys. Furthermore, the results are made even more credible by validating the scoring system by association with experimental binding affinities (such as IC?? or K? values). After docking, molecular dynamics (MD) simulations can be performed to improve binding poses, evaluate the ligand-receptor complex's temporal stability, and learn more about the dynamics of interactions. Virtual screening is used for large-scale drug discovery applications to test hundreds to millions of compounds against a single target protein. Batch processing of libraries such as ZINC or PubChem is made possible by high-throughput docking algorithms, which yield a prioritized list of possible hits according to their docking scores and binding energies.Following this screening process, promising candidates undergo lead optimization, which involves making chemical changes to enhance binding affinity, specificity, and pharmacokinetic characteristics including absorption, distribution, metabolism, excretion, and toxicity (ADMET). [72]The insights gained from docking serve as the basis for these modifications, and the modified molecules are repeatedly re-docked to assess the effects of each alteration. Lastly, it is crucial that all findings be **documented and reported. Along with the anticipated ADMET profiles, this entails summarizing important docking results, such as binding affinities, docking scores, and visual representations of important interactions. A comprehensive report serves as a foundational step in contemporary structure-based drug discovery and computational biology by guaranteeing transparency, reproducibility, and clarity in the communication of the molecular docking study's results.
CONCLUSION
Molecular docking has emerged as a cornerstone of computational drug discovery, allowing the possibility of predicting and analyzing molecular interactions between small molecules called ligands and biological targets, such as proteins and nucleic acids. The paper provides an overall perspective regarding the different parts included in a molecular docking protocol: protein and ligand preparation, homology modeling, docking methodologies, and post-docking analysis. Preparation of protein is crucial because it allows the accuracy of the structural and chemical properties of the receptor. The basis of accurate receptor modeling involves techniques such as energy minimization, hydrogen addition, and identification of the active site. Similarly, the ligand preparation is just as important, involving the optimization of geometry, conformations, and chemical states to resemble physiological conditions. Both of these steps ensure that, when done meticulously, significantly increase the reliability of docking predictions. Recent advancements in molecular docking tools and technologies have added precision and efficiency to the workflow. Advances in high-throughput virtual screening, flexible docking algorithms, and machine learning-augmented scoring functions enable the use of docking techniques in high-throughput mode on large compound libraries. Robust data resources, such as PDB, ZINC, and PubChem, offer the structural and chemical data needed for detailed docking studies. Modern tools like Auto Dock, Schrödinger Glide, and HADDOCK offer tailored solutions for diverse docking scenarios, including protein-ligand, protein-protein, and nucleic acid docking. The importance of homology modeling cannot be overstated because it allows researchers to construct accurate models for proteins lacking experimental structures. By combining these models with good grid generation and identification of the binding site, the reach of docking studies to understudied targets-such as orphan receptors or transient protein conformations-increases. Advances in scoring functions, interaction analysis tools, and molecular dynamics simulations improved the predictive power of docking studies. These tools provide an opportunity to refine binding poses, which gives better insight into molecular recognition, binding affinity, and stability. Moreover, the inclusion of docking results with biochemical validation experiments enhances confidence in the computational findings and bridges the gap between in-silico predictions and real-world applications. In conclusion, molecular docking continues to grow as an essential tool in modern drug discovery, structural biology, and chemical informatics. Such studies reveal insights into the identification of novel therapeutic agents, understanding disease mechanisms, and optimization of lead compounds. With state-of-the-art methodologies and computational power, molecular docking promises to accelerate drug discovery pipelines and pave the way for innovative therapeutic solutions. Future developments, including AI-driven docking and integration with multi-omics data, will undoubtedly push the boundaries of what can be achieved in this exciting and dynamic field.
REFERENCES

Dr. Rajaganapathy Kaliyaperumal*, Selvakumar R., Diana L., Aswin A., Keerthivasan K. M., Srinivasan. R., Advancing In-Silico Drug Discovery: A Comprehensive Strategy for Homology Modeling, Protein Preparation, and Ligand Optimization in Molecular Docking, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 5, 4338-4360. https://doi.org/10.5281/zenodo.15517725
 10.5281/zenodo.15517725
10.5281/zenodo.15517725