Aadhi Bhagawan College of Pharmacy, Rantham, Vembakkam T.K, Thiruvannamalai, Tamilnadu, India
Tedizolid Phosphate, a novel oxazolidinone antibiotic, has been widely studied using various advanced analytical methods to ensure its efficacy, stability, and safety in pharmaceutical formulations. This review highlights recent developments in liquid chromatography–mass spectrometry (LC-MS/MS), high-performance liquid chromatography (HPLC), reverse-phase HPLC (RP-HPLC), and spectrophotometric techniques for the qualitative and quantitative estimation of Tedizolid Phosphate. LC-MS/MS proved highly effective for identifying degradation products under ICH-recommended stress conditions. RP-HPLC using ?-cyclodextrin as a chiral selector demonstrated high enantiomeric resolution, while spectrophotometry based on hydrotropy offered a cost-effective alternative. HPLC methods were optimized for analyzing Tedizolid in biological matrices, ensuring robust detection and quantification. The study summarizes these techniques' validation parameters, including LOD, LOQ, recovery, and specificity, supporting their application in routine pharmaceutical analysis.
Formulation development is a key aspect of product development that can determine the patentability, lifecycle, and ultimately, the success of a pharmaceutical product. Companies integrate formulation development functions and personnel into their product development cycles in various ways. In fully integrated large pharmaceutical companies, specific departments may be dedicated to areas such as the physical characterization of drug substances and formulation-related issues. In many cases, other departments—often located in different buildings or locations—handle preclinical functions such as animal testing. These departments must collaborate closely to gain a comprehensive understanding of their unique drug substance.
Pharmaceutical formulation development serves as a bridge between the discovery of a new drug substance and the successful development of a commercial drug product. Formulation development scientists must determine the most appropriate route for achieving effective drug delivery based on patient needs, then optimize the formulation’s characteristics based on knowledge of the drug product’s bioavailability and processing requirements.
Formulation development encompasses a wide range of activities. Traditionally, it includes pre-formulation tasks such as analytical assay development and characterization, excipient screening to stabilize or enhance the solubility of the product, and dosage form development—whether it involves a solid, topical, aerosol, liquid, or lyophilized dosage form. Formulation development may also involve evaluating delivery options and assessing delivery device compatibility.
1.1 Analytical Development:
The purpose of analytical development is to establish the identity, purity, physical characteristics, and potency of drugs, including their bioavailability and stability. Analytical development plays a crucial role in demonstrating that analytical procedures are adequate for assessing drugs—particularly the active pharmaceutical ingredient (API).
Steps for Analytical Method Development:
Purpose of Analytical Method Development: In the pharmaceutical industry, analytical method development provides essential information on the potency, bioavailability, and stability of a drug, as well as its effects. The first step involves clearly defining the purpose of conducting analytical method development.
Documentation of Development Steps: In this step, all procedures and steps involved in method development are recorded in a laboratory notebook to ensure traceability and reproducibility.
Characterization of the Analyte: Both the biological and chemical properties, along with the physical characteristics of the analyte, are identified and documented. The analyte is then obtained and stored according to specific requirements. Suitable analytical techniques—such as chromatography, including High-Performance Liquid Chromatography (HPLC)—are selected for evaluation.
Definition of Method Requirements: The requirements for method development are defined and documented. All necessary materials, reagents, and instruments are procured to support the analytical process.
Literature Review and Assessment of Existing Methods: Relevant literature—including scientific journals, books, and publications—is reviewed to gather information about the analyte. This includes biological, chemical, and physical properties, as well as any previously established methods.
Selection of an Analytical Method: Based on the literature review, an appropriate analytical method is selected or adapted to meet specific requirements for the analyte. If no existing method is found, a new method is developed from scratch.
Instrument Setup: Analytical instruments are set up in accordance with their respective Standard Operating Procedures (SOPs). SOPs are standardized protocols that ensure consistent and accurate operation of laboratory instruments.
Optimization of the Method: The analytical method is optimized by systematically adjusting parameters to improve performance. This process follows a documented and structured approach, ensuring that each change is based on justified scientific reasoning.
Analytical Figures of Merit: Key performance characteristics—such as quantification limits, detection limits, analysis timeframe, operational cost, and sample preparation—are documented. These are known as the analytical figures of merit.
Evaluation of the Developed Method: The effectiveness of the developed method is evaluated to ensure it provides reliable and accurate identification of the analyte.
Sample Estimation, Quantification, and Analysis: The final step involves estimating and quantifying the analyte—such as a drug—in a matrix sample. This includes complete analysis and demonstration of method performance on real samples.
1.2 Analytical Technique:
Analytical technique is a method that is used to determine a chemical or physical property of a chemical substance, chemical element, or mixture. There are a wide variety of techniques used for analysis, from simple weighing to advanced techniques using highly specialized instrumentation.
A. Spectroscopy - Visible Spectroscopy, Ultraviolet Spectroscopy, Fluorimetry, Nephelometry, Turbidimetry, Atomic Absorption Spectroscopy, Infra-Red Spectroscopy, NMR Spectroscopy, ESR Spectroscopy, Mass Spectroscopy.
B. Chromatography - Column Chromatography, Ion-Exchange Chromatography, Gel-Permeation (Molecular Sieve) Chromatography, Affinity Chromatography, Paper Chromatography, Thin-Layer Chromatography, Gas Chromatography, Dye-Ligand Chromatography, Hydrophobic Interaction Chromatography, Pseudo-affinity Chromatography, High-Pressure Liquid Chromatography (HPLC)
C. Electrochemical Methods Of Analysis - Potentiometric Electrodes, Coulometric Methods, Voltammetry, Polarography, Stripping Voltammetry, Hydrodynamic Voltammetry, Amperometry.
D. Electrophoretic Methods - Capillary Electrophoresis (CE), Slab Electrophoresis, Gel Electrophoresis, Paper Electrophoresis, Immuno-electrophoresis, Zone Electrophoresis, Iso-electric focusing.
Fig: 1 Spectroscopy
1.3 Analytical Method Validation:
Analytical method validation is a critical process in pharmaceutical development. It confirms that the analytical methods used are suitable for their intended purpose and produce reliable, consistent, and accurate results. Validation ensures that the method meets regulatory requirements and scientific standards, supporting the safety, efficacy, and quality of pharmaceutical products.
The validation process typically involves assessing the following parameters:
Accuracy: Accuracy refers to the closeness of test results obtained by the method to the true value or a standard reference. It is usually determined by recovery studies using known concentrations of the analyte.
Precision: Precision is the degree of agreement among individual test results when the method is applied repeatedly. It includes:
Specificity (Selectivity): Specificity is the ability of the method to accurately measure the analyte in the presence of other components such as impurities, degradants, or matrix components.
Linearity: Linearity refers to the ability of the method to elicit results that are directly proportional to the concentration of analyte within a given range.
Range: The range is the interval between the upper and lower concentrations of analyte that the method can accurately and precisely measure.
Limit of Detection (LOD): LOD is the lowest amount of analyte in a sample that can be detected, but not necessarily quantified.
Limit of Quantitation (LOQ): LOQ is the lowest amount of analyte in a sample that can be quantitatively determined with acceptable precision and accuracy.
Robustness: Robustness measures the method’s capacity to remain unaffected by small, deliberate variations in method parameters (e.g., temperature, pH, flow rate), providing an indication of its reliability during normal usage.
System Suitability Testing (SST): SST evaluates whether the analytical system is functioning properly before and during sample analysis. Parameters such as resolution, repeatability, and column efficiency are typically assessed.
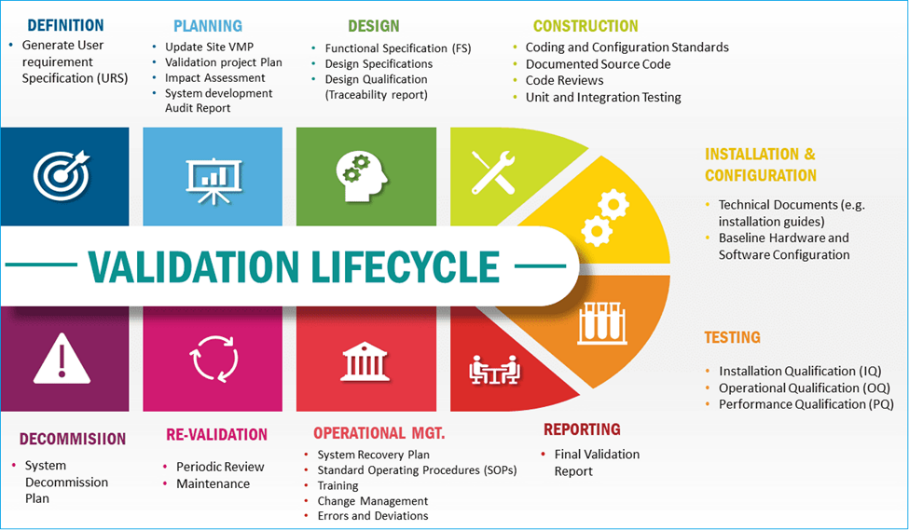
Fig: 2 Validation
DRUG NAME: TEDIZOLID PHOSPHATE:
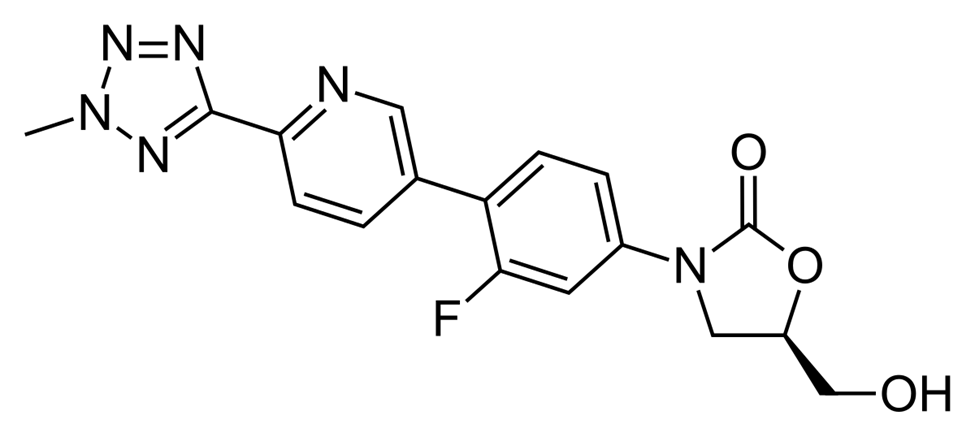
Fig: 3 Structure of Tedizolid Phosphate
Description:
Tedizolid phosphate is a phosphate monoester formed by the formal condensation of equimolar amounts of phosphoric acid with the hydroxy group of tedizolid. It acts as a prodrug of tedizolid and is used to treat acute bacterial skin infections caused by certain susceptible bacteria, including Staphylococcus aureus (both methicillin-resistant [MRSA] and methicillin-susceptible strains), various Streptococcus species, and Enterococcus faecalis. It is classified into various chemical groups, such as carbamate esters, organofluorine compounds, oxazolidinones, pyridines, tetrazoles, and phosphate monoesters.
Mechanism Of Action:
Tedizolid exhibits bacteriostatic activity by inhibiting bacterial protein synthesis through its binding to the 23s ribosomal RNA of the 50s subunit. Its chemical structure is similar to that of linezolid.both are synthetic molecule containing an oxazolidinone ring(ring A) and a lateral chain at the C5 position ,which enhance their activity against certain gram-positive bacteria and mycobacteria.
The primary chemical difference between the two compounds is that tedizolid contains a hydroxymethyl group in its side chain, which contributes to its activity against certain bacterial strains carrying the cfr gene. Additionally, tedizolid features a para-oriented D-ring structure, which increases its binding interactions with the peptidyl transferase center, thereby enhancing its potency compared to linezolid.
Pharmacodynamics:
The antimicrobial spectrum of tedizolid (TDZ) includes clinically significant Gram-positive bacteria, such as methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant S. aureus (MRSA), methicillin-susceptible S. epidermidis, methicillin-resistant S. epidermidis, vancomycin-sensitive and vancomycin-resistant enterococci (VRE), penicillin-susceptible Streptococcus pneumoniae (PSSP), penicillin-resistant S. pneumoniae (PRSP), and other commonly reported cutaneous and respiratory pathogens. Most of these Gram-positive bacteria are susceptible to TDZ, with minimum inhibitory concentration (MIC) values of ≤ 0.5 mg/L.
However, compared to its activity against Gram-positive organisms, TDZ shows reduced potency against Gram-negative bacteria, such as Haemophilus influenzae (MIC: 16 mg/L) and Moraxella catarrhalis (MIC: 4 mg/L). Similar to linezolid (LNZ), TDZ binds to the 50S subunit of bacterial ribosomal RNA and inhibits protein synthesis.
Pharmacokinetics:
Absorption: Tedizolid reaches peak plasma concentrations approximately three hours after oral administration and about one hour after intravenous administration. Its absolute oral bioavailability is around 91%, and food does not affect its absorption. When administered once daily, either orally or intravenously, tedizolid reaches steady-state concentrations within approximately three days.Following a single dose or at steady state, the maximum plasma concentration (Cmax) of tedizolid is 2.0 ± 0.7 mcg/mL and 2.2 ± 0.6 mcg/mL, respectively, for oral administration, and 2.3 ± 0.6 mcg/mL and 3.0 ± 0.7 mcg/mL, respectively, for intravenous administration. The time to reach maximum concentration (Tmax) has a median (range) of 2.5 hours (1.0–8.0) after oral administration and 3.5 hours (1.0–6.0) at steady state, while for intravenous administration, the median Tmax is 1.1 hours (0.9–1.5) after a single dose and 1.2 hours (0.9–1.5) at steady state.
The area under the concentration-time curve (AUC) is 23.8 ± 6.8 mcg·hr/mL after a single oral dose and 25.6 ± 8.4 mcg·hr/mL at steady state, compared to 26.6 ± 5.2 mcg·hr/mL and 29.2 ± 6.2 mcg·hr/mL, respectively, for intravenous administration.
Volume of distribution: The volume of distribution for tedizolid after a single 200 mg intravenous dose ranges from 67 to 80 liters. In a study involving oral administration of 200 mg tedizolid at steady state, the volume of distribution was reported to be 108 ± 21 liters, whereas a single 600 mg oral dose resulted in an apparent volume of distribution of 113.3 ± 19.3 liters. Tedizolid has been shown to penetrate the interstitial space of both adipose and skeletal muscle tissues and is also present in the epithelial lining fluid and alveolar macrophages.
Protein binding: Approximately 70% to 90% of tedizolid binds to human plasma proteins.
Metabolism: Tedizolid is administered as a phosphate prodrug, which is converted into the active circulating form, tedizolid. Before excretion, the majority of tedizolid is metabolized in the liver into an inactive sulfate conjugate; however, this process is unlikely to involve cytochrome P450 enzymes.
Route of elimination: Following a single oral dose, approximately 82% of tedizolid is excreted in the feces and 18% in the urine. The majority is eliminated as the inactive sulfate conjugate, with only about 3% recovered unchanged. More than 85% of the elimination takes place within 96 hours.
Half-life: The half-life of tedizolid is approximately 12 hours.
Clearance: Tedizolid exhibits an apparent oral clearance of 6.9 ± 1.7 L/hr following a single dose and 8.4 ± 2.1 L/hr at steady state. Its systemic clearance is 6.4 ± 1.2 L/hr after a single dose and 5.9 ± 1.4 L/hr at steady state.
Side effects: Severe stomach pain, diarrhoea that is watery or bloody (even if it occurs months after your last dose)
Toxicity: Toxicity information for tedizolid is limited. In cases of overdose, patients may be at increased risk of experiencing severe adverse effects, including nausea, headache, dizziness, diarrhea, and vomiting. Symptomatic and supportive care is recommended.
Drug interaction: Both linezolid and tedizolid are weak inhibitors of monoamine oxidase (MAO), which can theoretically result in interactions with serotonergic medications, such as selective serotonin reuptake inhibitors (SSRIs). However, tedizolid poses a lower risk of inducing serotonin syndrome compared to linezolid, making it a safer option for patients taking these drugs.
3.1 LIQUID CHROMATOGRAPHY–MASS SPECTROMETRY (LC-MS/MS):
LC-MS/MS is an advanced analytical technique combining the separation power of liquid chromatography with the detection specificity and sensitivity of mass spectrometry. It is extensively used in pharmaceutical analysis for identifying and characterizing drug degradation products, studying impurity profiling, and validating drug stability under stress conditions as per ICH guidelines.
Table: 1 Liquid Chromatography–Mass Spectrometry (LC-MS/MS)
|
S.No |
Stationary phase / Instrumentation |
Method/Conditions |
Results |
Reference |
|
1 |
UPLC–MS? and LC-HRMS |
Forced degradation under hydrolytic (acid, base, neutral), oxidative, photolytic, thermal conditions (ICH Q1A(R2)) |
Drug degraded under acid, base, oxidative conditions |
K.L. Leach et: al.(17) |
*UPLC–MS? – Ultra Performance Liquid Chromatography–Multistage Mass Spectrometry
*LC-HRMS – Liquid Chromatography–High-Resolution Mass Spectrometry
*ICH Q1A(R2) – International Guidelines for Stability Testing of New Drug Substances and Products.
3.2 NANTIOSEPARATION OF TEDIZOLID PHOSPHATE BY RP-HPLC:
A reversed-phase high-performance liquid chromatography (RP-HPLC) method was developed using B-cyclodextrin (B-CD) as a chiral mobile phase additive for the enantiomeric separation and quantification of the S-enantiomer of Tedizolid phosphate. The method was optimized by varying mobile phase composition, pH, and column temperature to achieve satisfactory resolution.
Table: 2 Nantioseparation Of Tedizolid Phosphate By RP-HPLC
|
Sr. No. |
Stationary phase |
Mobile phase |
Flow rate / Detection / Retention time |
Results |
Reference |
|
1 |
Phenomenex Luna, Phenyl-Hexyl |
Buffer (pH 7.0) of disodium hydrogen phosphate with B-cyclodextrin, triethylamine, and acetonitrile |
Column temp: 20°C |
LOD: 0.10 µg/mL |
Bressolle et:al (20)
|
3.3 SPECTROPHOTOMETRIC METHOD FOR TEDIZOLID PHOSPHATE BY HYDROTROPY:
This spectrophotometric method is based on the principle of hydrotropy to enhance the solubility of Tedizolid Phosphate for its accurate and cost-effective estimation. Sodium benzoate was used as the hydrotropic agent, improving the drug's aqueous solubility and enabling its estimation at a specific wavelength.
Table: 3 Spectrophotometric Method For Tedizolid Phosphate By Hydrotropy
|
Sr. No. |
Method Principle / Reagent |
Conditions |
Results |
Reference |
|
1 |
Spectrophotometry |
Wavelength: Not specified |
Method is simple, accurate, robust |
Singh A, et:al |
3.4 HIGH PERFORMANCE LIQUID CHROMATOGRAPHY (HPLC):
High Performance Liquid Chromatography (HPLC) is a powerful and reliable analytical method commonly used in pharmaceutical analysis to determine the presence and concentration of various compounds. In this study, a simple and reliable HPLC method was developed and validated to analyze tedizolid in human plasma, human serum, saline, and CD-1 mouse plasma. An ultraviolet detector set at 251 nm was used with a reverse phase column. The mobile phase consisted of sodium acetate, deionized water, and acetonitrile with a flow rate of 1.0 ml/min. 4-nitroaniline was used as the internal standard. All chromatographic procedures were performed at room temperature.
Table: 4 High Performance Liquid Chromatography (HPLC)
|
Sr. No |
Stationary phase |
Mobile phase |
Flow rate, method of detection, retention time |
Results |
Reference |
|
1 |
Reverse Phase Column |
Sodium acetate buffer (0.0192 M): Acetonitrile (23%) |
1.0 ml/min, UV at 251 nm, Not specified |
Internal standard: 4-nitroaniline |
Santini D.A. et:al
|
High Performance Liquid Chromatography (HPLC) is a widely used technique in pharmaceutical analysis for quantifying drug concentrations in biological matrices. In this study, a high-performance liquid chromatography-fluorescence detector assay was developed to measure tedizolid (TZD) concentration in human serum. The chromatographic separation was performed on a 5 μm octadecyl silane hypersil column (150 mm × 4.6 mm). The mobile phase consisted of 0.1 M phosphoric acid and methanol in the ratio of 60:40, adjusted to pH 7.0. Detection was performed at 300 nm and 340 nm for excitation and emission, respectively. The retention time for TZD and internal standard was found to be 12.9 and 8.8 min. High linearity was observed within the concentration range of 0.025–10.0 μg/mL. The accuracy of TZD quantification ranged from 99.2% to 107.0%. The method showed excellent extraction recoveries and reliable detection limits.
Table: 5 HPLC Of Tedizolid Phosphate
|
Sr. No |
Stationary phase |
Mobile phase |
Flow rate, method of detection, retention time |
Results |
Reference |
|
1 |
Octadecyl Silane Hypersil (150 mm × 4.6 mm, 5 μm) |
0.1 M Phosphoric acid : Methanol (60:40, pH 7.0) |
Excitation 300 nm, Emission 340 nm, Retention time: 12.9 min (TZD), 8.8 min (IS) |
LOD: 0.01 μg/mL, LOQ: 0.025 μg/mL, Recovery: 100.4% to 114.1% |
Tsuji et:al |
Table: 6 HPLC Of Tedizolid Phosphate
|
Sr. No |
Stationary Phase |
Mobile Phase |
Flow rate Method of detection Retention time |
Result |
References |
|
1 |
Silica gel 60 F254 (10 cm × 10 cm with a 250 µm layer thickness) |
Acetone, Methanol, Toluene and Formic acid (4:3:2:1, v/v/v/v) |
Retention factor 0.61 |
LOD-1074.928 ng/spot LOQ-3257.54 ng/spot |
Suresh Kumar et al. |
|
2 |
Credchrom C18 column (250 mm × 4.6 mm × 5 µm) |
Phosphate buffer and aceto nitrile (80:20 v/v) |
1.0 ml/min UV at 241 nm |
LOD-0.577722 µg/ml LOQ-1.750673 µg/ml |
Vaibhav S. Adhao et al. |
|
3 |
BRISALC2 C18 (25 mm × 0.46 mm, 5 µm) |
Methanol:phosphate buffer (10 mM) (80:20 v/v) |
1.0 ml/min UV at 243 nm |
LOD-0.137 µg/ml LOQ-1.417 µg/ml |
Sharanabasava Navali et al. |
|
4 |
Phenomenx C18 (250mm × 4.6 mm, 5 µm) |
Methanol:0.05% TFAA in water (20:80 v/v) |
1.0 ml/min UV at 240 nm |
LOD- 3.3 σ/s LOQ-10 σ/s |
Hemant Chikhale et al. |
|
5 |
Purospher star RP18 endcapped (250 mm × 4.6 mm, 5 µm) |
Water:Methanol (80:20 v/v) |
0.8 ml/min UV at 234 nm |
- |
Ajay Sanjay Salvi et al. |
|
6 |
Hypersil gold ODS endcapped column (150 × 4.6 mm, 3 micron) |
Water:Acetonitrile (15:85 v/v) |
1.0 ml/min UV at 240 nm |
LOD- 3.3 σ/s LOQ-10 σ/s |
Avnish Jain et al. |
DISCUSSION:
The analytical landscape for Tedizolid Phosphate demonstrates a well-rounded approach to ensuring drug quality and compliance with regulatory standards. The LC-MS/MS technique, as reported by Leach et al. (17), remains the gold standard for stability testing, allowing detection of degradation products under acid, base, oxidative, photolytic, and thermal conditions. This method’s high resolution and sensitivity support its use in forced degradation and impurity profiling.
The enantioselective RP-HPLC method developed by Bressolle et al. (20) offers high specificity using β-cyclodextrin as a chiral selector. With recovery rates between 96.9%–105.3%, an R² value of 0.998, and LOD/LOQ of 0.10 µg/mL and 0.30 µg/mL respectively, this method is both precise and reliable for separating Tedizolid enantiomers—critical for evaluating pharmacokinetics and toxicity profiles.
A cost-effective spectrophotometric method using sodium benzoate as a hydrotropic agent by Singh et al. enables routine estimation in formulations. Its simplicity, linearity (10–50 µg/mL), and robust performance make it ideal for laboratories lacking sophisticated instrumentation.
Several HPLC methods have been developed for Tedizolid detection in biological samples such as human plasma, serum, saline, and mouse plasma. These methods use internal standards like 4-nitroaniline and exhibit high recovery rates and excellent linearity. Tsuji et al. notably used a fluorescence detector, achieving LOD as low as 0.01 µg/mL and LOQ of 0.025 µg/mL, with recoveries exceeding 100%.
Other HPLC methods (Santini D.A. et al.; Vaibhav S. Adhao et al.; Suresh Kumar et al.) use various C18 and silica-based columns with different mobile phases, retention times, and detection modes (UV, fluorescence) for Tedizolid analysis, demonstrating the versatility and adaptability of HPLC platforms.
Each analytical approach brings unique strengths, with method validation in accordance with ICH guidelines—ensuring accuracy, precision, specificity, robustness, and reproducibility across formulations and biological matrices.
CONCLUSION:
Multiple validated analytical methods for Tedizolid Phosphate have been developed, ranging from advanced LC-MS/MS techniques for impurity profiling to robust and cost-effective spectrophotometric methods for routine estimation. RP-HPLC has shown high specificity for enantiomeric separation, while several HPLC protocols provide accurate and sensitive drug quantification in biological matrices. These methods, adhering to ICH validation guidelines, are crucial for supporting drug development, regulatory compliance, and quality control of Tedizolid formulations. Continuous method optimization will enhance their applicability across research and clinical environments.
REFERENCES

Dr. K. Kaveri, B. Jana, R. Jeevitha, L. Jayabharath, C. Jana, N. Jaisuriyan, Analytical Methods for Estimation of Tedizolid Phosphate in Pharmaceutical Dosage Form – A Review, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 3248-3258. https://doi.org/10.5281/zenodo.16410515
 10.5281/zenodo.16410515
10.5281/zenodo.16410515
