
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of pharmaceutical chemistry, College of Pharmaceutical Sciences, Government Medical College, Thiruvananthapuram.
Benzimidazole derivatives constitute an important class of heterocyclic compounds with significant pharmaceutical relevance. Their unique structural framework, combining the fused benzene and imidazole rings, enables diverse interactions with biological targets, resulting in a broad spectrum of pharmacological activities. These include antimicrobial, antiviral, anticancer, anti-inflammatory, antiulcer, antidiabetic and antihypertensive effects. Structural modifications at different positions on the benzimidazole core have been shown to greatly influence potency, selectivity, and pharmacokinetic profiles. Recent research has focused on synthesizing novel derivatives through conventional and green chemistry approaches, exploring their structure–activity relationships (SAR), and evaluating their potential as lead molecules in drug discovery. This review summarizes the chemical diversity, pharmacological applications, and SAR trends of benzimidazole derivatives, highlighting their versatility and potential in the development of new therapeutic agents.
Benzimidazole is a heteroaromatic bicyclic system that results from the fusion of a benzene ring with an imidazole ring. The presence of two nitrogen atoms at the 1 and 3 positions within the imidazole moiety imparts remarkable versatility to this scaffold in the field of medicinal chemistry. Owing to its structural features, the benzimidazole core is considered a “privileged scaffold”, capable of binding to diverse biological targets through hydrogen bonding, π–π interactions, and hydrophobic contacts1. This heterocyclic framework is naturally found in biomolecules such as vitamin B?? (cobalamin), where benzimidazole functions as a coordinating ligand, underscoring its biological significance. Because of its structural similarity to purines, benzimidazole derivatives readily interact with nucleic acids and enzymes, which accounts for their broad range of pharmacological activities2. In recent decades, benzimidazole derivatives have been shown to possess a wide array of pharmacological properties, such as antimicrobial, anti-inflammatory, anticancer, antiviral, anti-ulcer, antihypertensive, and antiparasitic activities. Structural modifications, particularly at the 2, 5, and 6 positions of the benzimidazole ring, have been found to significantly improve biological efficacy, thereby establishing this nucleus as a promising scaffold for drug design and development3. At present, numerous benzimidazole-based drugs, including albendazole, mebendazole, omeprazole, and thiabendazole, are widely used in clinical practice, demonstrating their therapeutic relevance. Ongoing efforts in synthetic modification, molecular docking, and structure–activity relationship (SAR) investigations continue to advance benzimidazole research, paving the way for the development of safer and more effective therapeutic agents in medicinal chemistry4.
Benzimidazole Derivatives as Anti-Inflammatory Agents
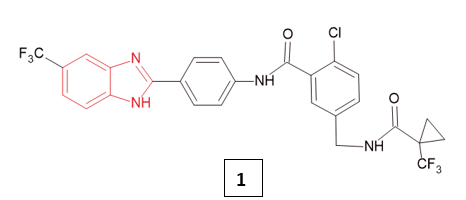
Current treatment strategies for inflammatory diseases mainly focus on blocking the production or activity of mediators responsible for the body’s response to injury5. The management of pain and inflammatory conditions typically follows a stepwise approach, involving the use of conventional non-steroidal anti-inflammatory drugs (NSAIDs), selective cyclooxygenase-2 (COX-2) inhibitors, as well as corticosteroids and immunosuppressive agents6. Azize Gizem Ergül et al (2024) investigated benzimidazole derivatives as inhibitors of microsomal prostaglandin E2 synthase-1 (mPGES-1), a key enzyme in the arachidonic acid (AA) cascade, a pathway strongly linked to inflammation. Prostaglandin E2 (PGE2) is produced through the cyclooxygenase (COX) pathway, involving COX-1/2 isoforms and three prostaglandin E synthases (mPGES-1, mPGES-2, and cPGES). Among these, mPGES-1 is the major contributor to pro-inflammatory PGE2 synthesis, with its expression markedly elevated during inflammation, often in parallel with COX-2. Because of its central role, selective inhibition of mPGES-1 has been proposed as a promising alternative to conventional NSAIDs, as it may specifically block the pathological overproduction of PGE2 without suppressing other prostaglandins and thromboxanes, thereby potentially avoiding the gastrointestinal and cardiovascular risks commonly associated with NSAIDs. Compound 1, exhibited exceptional potency and selectivity for mPGES-1 (IC50 = 2.9 nM), with no effect on COX-1, COX-2, 5-LOX, or FLAP in vitro. It acted in a noncompetitive, reversible manner, independent of PGH2 substrate levels. Further cellular studies in human monocyte-derived macrophages and whole blood confirmed that compound 1 effectively suppressed PGE2 production under inflammatory conditions, while leaving other COX/LOX-derived lipid mediators unaffected a distinct advantage over NSAIDs. In preclinical guinea pig models, compound 1 significantly reduced fever, pain, and inflammation. At 30 mg/kg, it provided analgesic effects comparable to indomethacin, but with superior anti-inflammatory activity, markedly decreasing carrageenan-induced paw edema and hyperalgesia. In an LPS-induced pyresis model, compound 1 displayed potent and sustained antipyretic activity, even surpassing indomethacin at later time points (180 and 240 minutes). Importantly, safety evaluations indicated only mild alterations in liver enzyme markers and histopathology compared to indomethacin, suggesting a more favourable hepatotoxicity profile. Overall, these findings highlight benzimidazole-based mPGES-1 inhibitors, particularly compound 1, as promising candidates for safer and more selective anti-inflammatory therapy7.
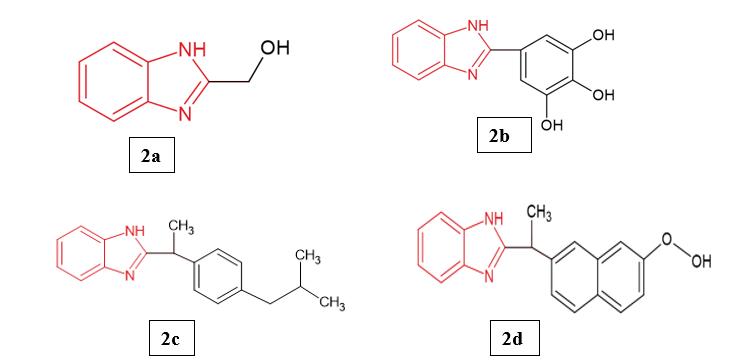
Saikat Kumar Poddar et al. (2025) explored the therapeutic potential of newly synthesized benzimidazole derivatives by designing and obtaining four mono-substituted analogs (2a–2d) in good yields (66–94%). The biological evaluation encompassed in vivo assays for analgesic, anti-inflammatory, and antidiarrheal activities, alongside in silico docking studies against key targets including COX-1, COX-2, and the κ-opioid receptor. Among the compounds, 2d emerged as the most active analgesic, achieving 62.37% writhing inhibition at 50 mg/kg, with 2b (58.98%) and 2c (57.29%) showing moderate efficacy relative to diclofenac sodium (92.54%). In anti-inflammatory testing, 2c and 2d demonstrated remarkable inhibition of paw edema (80.61% and 88.30%, respectively), notably exceeding the standard diclofenac sodium (56.97% in the first hour). For antidiarrheal effects, 2a was the most effective, inhibiting defecation by 66.67% at 50 mg/kg, approaching the activity of loperamide (75.0% at 25 mg/kg), whereas other analogs were less significant. Docking analyses supported the pharmacological findings, with 2d exhibiting the highest binding affinities toward COX-1 (−8.7 kcal/mol) and COX-2 (−8.9 kcal/mol), outperforming diclofenac sodium (−8.2 and −8.4 kcal/mol). The study highlights that substituent bulkiness on the benzimidazole nucleus strongly influences COX enzyme affinity, suggesting that these derivatives act primarily via COX inhibition8.
Benzimidazole Derivatives as Anticancer Agents
Cancer, the second leading cause of death worldwide, arises from deregulated cell cycles causing uncontrolled growth and loss of differentiation. To address resistance issues, novel heterocyclic compounds are being explored, as they offer unique reactivity, allow bioactive substitutions, and provide advantages such as eco-friendly synthesis, selectivity, high yields, and cost-effectiveness9.
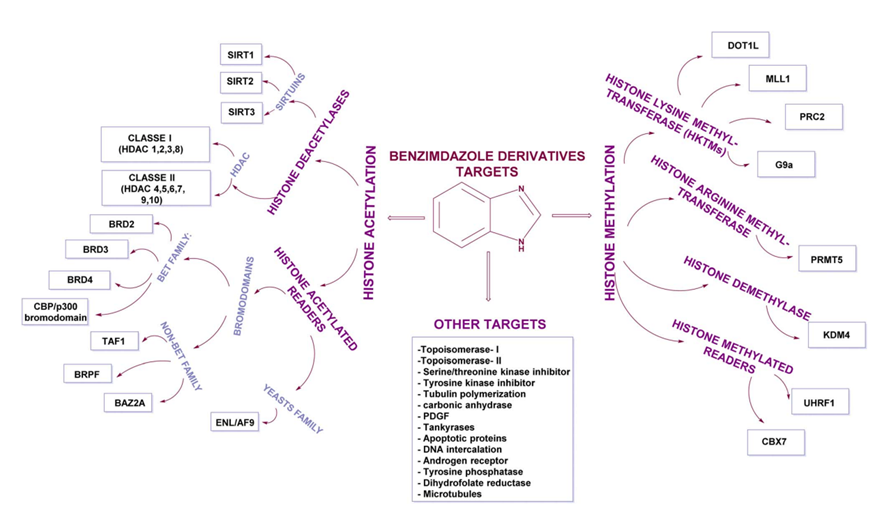
Figure 1: Representation of various targets of 1H-benzimidazolederivatives having anticancer effects10.
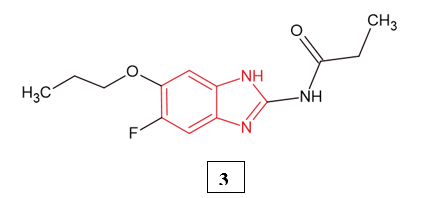
B. Koh et al. (2024) evaluated the benzimidazole derivative compound 3 as a potential therapy for platinum-resistant ovarian cancer. Using both chemo-sensitive (A2780, HeyA8, SKOV3ip1) and chemo-resistant (A2780-CP20, HeyA8-MDR, SKOV3-TR) epithelial ovarian cancer cell lines, they assessed proliferation (MTT assay), apoptosis (Annexin-V flow cytometry), and cell cycle effects. In vivo, compound 3 was orally administered in orthotopic mouse and patient-derived xenograft (PDX) models, with tumor growth, body weight, tumor burden, and nodule counts evaluated. Immunohistochemistry (MCM2, TUNEL) and molecular studies, including docking and tubulin polymerization assays, were conducted to explore the mechanism. Compound 3 inhibited proliferation in a dose- and time-dependent manner, enhanced apoptosis, and induced G2/M cell cycle arrest most prominently in resistant cells. Docking studies revealed strong binding at the colchicine site of tubulin, confirmed by tubulin destabilization activity. In vivo, compound 3 markedly suppressed tumour growth without systemic toxicity, with treated tumours showing reduced proliferation and increased apoptosis11.
Othman et al. (2023) focused on designing novel benzimidazole–triazole hybrids as multi-target anticancer agents against EGFR, VEGFR-2, and Topo II. Among the synthesized compounds, 4a and 4b showed the strongest activities across HepG-2, HCT-116, MCF-7, and HeLa cell lines. Compound 4a displayed remarkable potency, with EGFR inhibition close to Gefitinib, stronger Topo II inhibition than Doxorubicin, and moderate VEGFR-2 inhibition. Compound 6g was also active but generally less potent than 4a. Both compounds exhibited reduced toxicity toward normal WI-38 cells and favorable selectivity compared to Doxorubicin. Mechanistic studies revealed that 4a intercalated DNA and induced S-phase arrest, while 4b bound DNA and caused G2/M arrest, both triggering apoptosis. Docking studies further supported their strong interactions with EGFR, VEGFR-2, and Topo II active sites12.
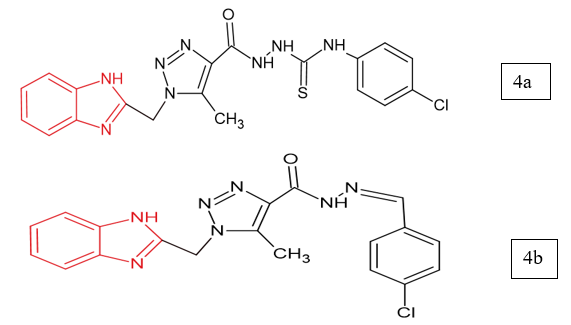
Benzimidazole Derivatives as Anti-Ulcer Agents
Peptic ulcer disease remains a major health concern worldwide, particularly in developing countries. The identification of H+/K+ ATPase as the key gastric proton pump has led to significant interest in its inhibition for gastric acid control. This paved the way for the development of benzimidazole sulfoxide–based antisecretory drugs. Timoprazole was the first well-characterized proton pump inhibitor, later followed by more effective agents such as picoprazole and omeprazole. Owing to their strong biological activity and broad therapeutic potential, synthetic benzimidazole derivatives continue to play an important role in managing various pathological conditions13.
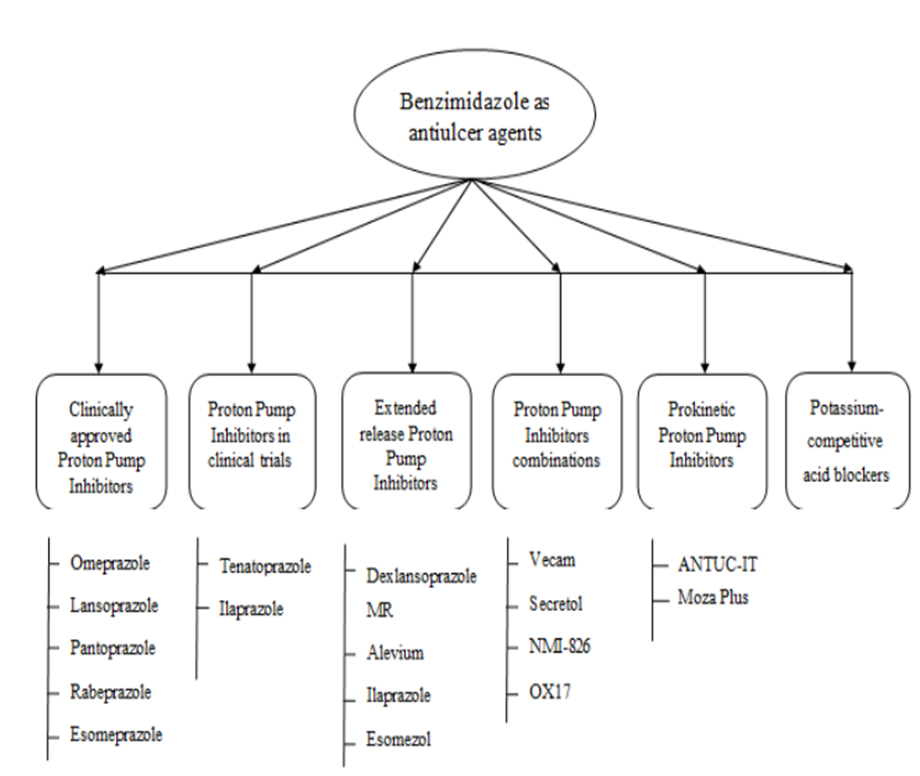
Figure 2: Benzimidazole derivatives as antiulcer agents.
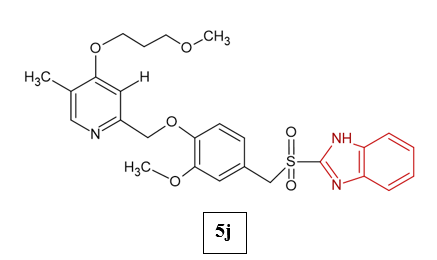
Radhamanalan et al. (2018) synthesized twelve racemic-substituted benzimidazoles (5a–l) via selective sulfide oxidation to sulfoxides and evaluated them for antiulcer potential using in vitro and in silico methods. All compounds showed strong, dose-dependent inhibition of H?/K?-ATPase, comparable to omeprazole, with compound 5j emerging as the most potent (IC?? = 0.025 μM; 86.12% activity vs. omeprazole’s 82.54%). Docking studies supported their strong binding to H?/K?-ATPase (binding energies −8.94 to −32.88 kcal/mol; ki = 0.027–2 μM). Safety assessment using HRBC membrane protection assays confirmed low toxicity, with 5d, 5i, and particularly 5j showing high stabilization (up to 98.45%)14.
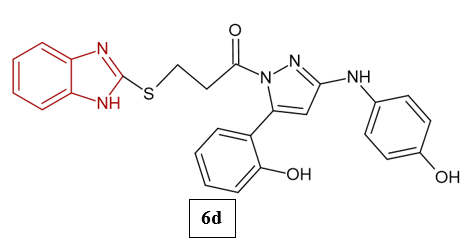
Noor et al. (2017) synthesized and characterized six novel benzimidazole–pyrazole hybrids (6a–6f) and evaluated their anti-ulcer activity using an ethanol-induced gastric ulcer model in Albino rats. All compounds showed stronger anti-ulcer effects than omeprazole, achieving 72–83% inhibition at just 500 µg/kg, compared to omeprazole’s 83% at 30 mg/kg. Among them, compound 6d was the most effective, with 83.1% inhibition and the highest binding affinity (−9.8 kcal/mol) to H?/K?-ATPase13.
Benzimidazole Derivatives as Antidiabetic Agents
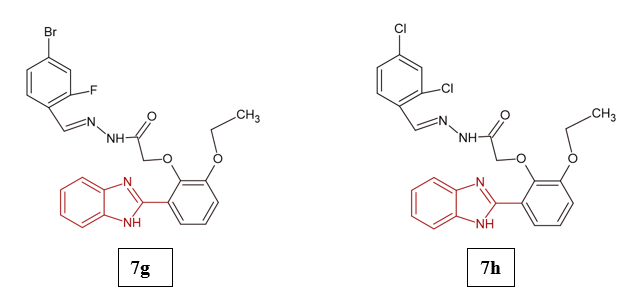
Type 2 diabetes mellitus (T2DM) is a long-term metabolic condition marked by persistent elevation of blood glucose levels. Factors such as obesity, sedentary lifestyle, and oxidative stress are considered major contributors to its development. If left unmanaged, T2DM can lead to severe complications, including cardiovascular disorders, retinopathy, neuropathy, and nephropathy. According to the International Diabetes Federation (IDF), around 425 million people worldwide were affected by diabetes in 2017, and this number is projected to rise to 629 million by 2045. In glucose metabolism, the enzymes α-amylase and α-glucosidase play crucial roles, as their activity contributes to hyperglycemia. Therefore, inhibiting these enzymes has emerged as an effective strategy for controlling T2DM14. Abdul Shakoor et al. (2024) investigated the anti-diabetic properties of Schiff bases incorporating a benzimidazole framework. Their findings showed that six derivatives (7g, 7h, 7q, 7p, 7i, and 7m) exhibited significant inhibitory effects on α-amylase (IC??: 2.15 ± 0.17–15.18 ± 3.29 µM) and α-glucosidase (IC??: 4.21 ± 0.78–16.10 ± 0.52 µM), outperforming the reference drug acarbose (IC??: 16.06 ± 0.05 µM for α-amylase; 16.65 ± 0.07 µM for α-glucosidase). Among them, compounds 7g and 7h demonstrated the strongest dual inhibitory activity. The study also highlighted that Schiff bases containing electron-withdrawing substituents displayed enhanced anti-diabetic potential. Results from time-dependent density functional theory (TD-DFT) indicated that 7g and 7h possessed lower electrophilicity and greater bioactivity than 7i and 7m. Furthermore, molecular dynamics simulations confirmed the stability of the protein–ligand complexes, while cytotoxicity assays revealed that these compounds were non-toxic to HepG2 cells at the tested doses15.
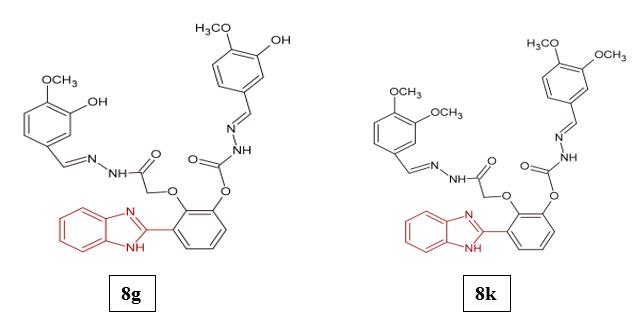
Hamid Ullah et al. investigated the anti-diabetic potential of newly synthesized benzimidazole-based bis-Schiff base derivatives, with emphasis on their in vitro inhibitory activity against α-amylase and α-glucosidase. The results revealed that seven compounds showed remarkable enzyme inhibition, with IC?? values between 2.84 ± 0.12 and 16.70 ± 1.23 µM, surpassing the standard drug acarbose (IC?? = 16.08 ± 0.18 and 16.15 ± 0.04 µM). Among them, compound 8k was identified as the most effective dual inhibitor of both α-glucosidase and α-amylase, displaying IC?? values of 2.84 ± 0.12 µM and 3.16 ± 0.05 µM, respectively. Compound 8g also exhibited notable α-glucosidase inhibition (IC?? = 6.94 ± 0.07 µM). Interestingly, these derivatives showed considerably higher plasma protein binding (98.347–98.894%) compared to acarbose (27.668%), which may lower the free drug fraction and thus affect in vivo efficacy. Structure–activity relationship (SAR) studies highlighted that electron-donating groups like methoxy and hydroxyl, particularly at the meta and para positions of the benzene ring, played a key role in enhancing enzyme inhibitory activity16.
Benzimidazole Derivatives as Antimicrobial and Antiviral Agents
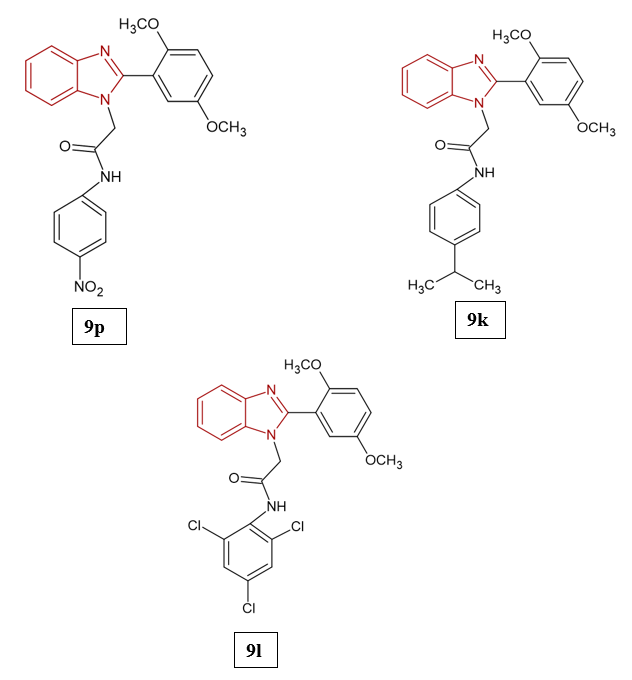
Antimicrobial and antiviral agents play a crucial role in modern medicine, as infectious diseases caused by bacteria, fungi, and viruses remain a major global health concern. The rise of multidrug-resistant pathogens, including Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, and Candida species, has intensified the search for new and more effective antimicrobial therapies17. Antimicrobial agents exert their effects through multiple mechanisms, including the inhibition of cell wall formation, interference with protein or nucleic acid synthesis, and disruption of membrane integrity18. In a similar manner, antiviral drugs act by blocking key steps in the viral life cycle such as attachment, genome replication, protein processing, and viral assembly, thereby suppressing infection19. A wide range of therapeutics, including nucleoside and non-nucleoside analogues, polymerase inhibitors, and protease inhibitors, have been successfully employed in the management of diseases such as HIV, hepatitis B and C, influenza, and more recently, coronavirus infections. Nevertheless, the growing problem of drug-resistant microbes and the emergence of new viral pathogens highlight the urgent need for continued exploration and development of novel antimicrobial and antiviral strategies20. Mohapatra et al. (2025) designed and synthesized 17 benzimidazole-based derivatives to evaluate their potential as NNRTIs for HIV and as antimicrobial agents. Structural modifications at the C-2 and N-1 positions optimized hydrophilic and hydrophobic balance for improved target binding. Biological screening revealed compound 9p as the most potent anti-HIV agent, showing 88.08% inhibition of HIV-1 reverse transcriptase with a docking score of −6.3 kcal/mol, attributed to nitro and chloro substituents on the third phenyl ring. Compound 9k demonstrated strong anti-mycobacterial activity against Mycobacterium smegmatis (MIC 0.195 μg/mL) and broad-spectrum antibacterial activity against S. aureus, B. subtilis, E. coli, and P. aeruginosa (MIC 3.125–6.25 μg/mL), due to nitro and methoxy substitutions. Additionally, compound 9l showed the best antifungal activity, with MICs of 6.25 μg/mL against C. albicans and 3.125 μg/mL against A. niger21.
Benzimidazole Derivatives as Antihypertensive Agents
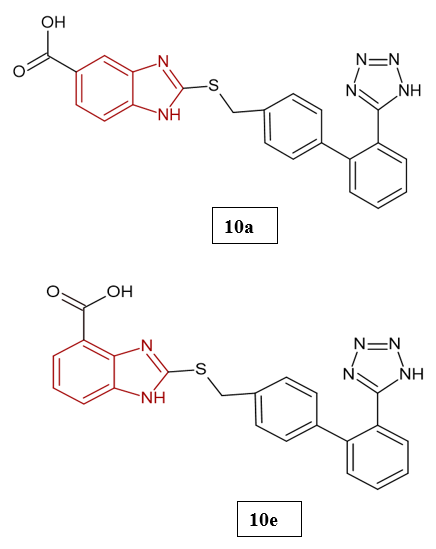
Hypertension is a major global health concern and a leading contributor to cardiovascular disease, stroke, and renal complications. Despite the wide range of available therapies, the growing prevalence of resistant hypertension underscores the need for more potent and safer treatment options. Current antihypertensive agents act through different mechanisms, including suppression of the renin–angiotensin–aldosterone system (RAAS), calcium channel inhibition, β-adrenergic receptor blockade, and diuretic action22. In drug development, heterocyclic ring systems have been particularly valuable, providing structural diversity and enhanced pharmacokinetic properties. Within this context, the benzimidazole ring has emerged as an important pharmacophore in antihypertensive drug design due to its strong interaction with the angiotensin II type 1 (AT?) receptor. Several widely used angiotensin II receptor blockers (ARBs), including losartan, candesartan, and telmisartan, contain a benzimidazole core, which is key to their high receptor affinity, selectivity, and favorable pharmacological profiles23. Abraham Gutiérrez-Hernández et al. (2024) reported the synthesis of seven benzimidazole derivatives, with their structures confirmed through spectroscopic analyses. Biosimulation studies indicated that compounds 10a–10g were computationally safe hits, displaying favourable pharmacodynamic and biopharmaceutical profiles, with predicted AT? Ang II receptor antagonism and minimal cardiotoxic risk. Among them, compounds 10a and 10e showed the greatest potential in silico. Ex vivo assays revealed that compound 10a exerted a dual vasorelaxant effect, functioning both as an insurmountable AT? receptor antagonist and as a partial inhibitor of the L-type CaV1.2 calcium channel. Molecular docking further clarified the binding interactions of compound 10a within both targets, supporting its multitarget mechanism. In vivo evaluation using a spontaneously hypertensive rat (SHR) model demonstrated that intraperitoneal administration of compound 10a (20 mg/kg) reduced systolic blood pressure by 25%, validating its ex vivo vasorelaxant and multitarget antihypertensive activity. The study emphasized the critical role of the biphenyl moiety in compound 10a and highlighted the consistency between computational predictions and experimental findings, underscoring the effectiveness of rational drug design in developing multitarget antihypertensive agents24.
CONCLUSION
Benzimidazole derivatives have emerged as a highly versatile scaffold in medicinal chemistry, recognized for their structural flexibility and extensive range of therapeutic applications. They exhibit notable efficacy as antimicrobial, antiviral, anticancer, anti-inflammatory, antidiabetic, antihypertensive, and gastroprotective agents, with several derivatives successfully advanced into clinical practice. Structure–activity relationship insights suggest that even minor modifications to the benzimidazole framework can significantly influence biological potency, improve selectivity, and optimize pharmacokinetic behavior. Although challenges such as resistance and safety remain to be addressed, ongoing exploration of benzimidazole chemistry continues to open avenues for the development of safer and more effective drug candidates. Collectively, the benzimidazole scaffold remains an indispensable platform in drug design, offering vast opportunities to tackle current and emerging health challenges.
Future Scope
The continued exploration of benzimidazole derivatives offers immense potential in modern drug discovery. Future research should focus on rational design strategies, integrating computational modeling, molecular docking, and advanced structure–activity relationship (SAR) studies to optimize both potency and selectivity. The development of multitarget-directed ligands (MTDLs) based on the benzimidazole scaffold could provide effective therapeutic options against complex and multifactorial diseases such as cancer, diabetes, neurodegenerative disorders, and cardiovascular disorders. Furthermore, nanotechnology-based drug delivery systems can enhance the bioavailability and targeted delivery of benzimidazole derivatives, thereby reducing systemic toxicity. With the growing challenge of antimicrobial and antiviral resistance, benzimidazole chemistry also holds promise for designing next-generation therapeutics with novel mechanisms of action. In addition, exploration of green chemistry and sustainable synthetic methodologies will further expand the accessibility of diverse benzimidazole analogues. Overall, the future of benzimidazole research lies in utilizing its chemical versatility to address unmet clinical needs and to deliver safer, more effective, and patient-friendly therapeutics.
REFERENCES


Aiswarya S.*, Sherin A. Hameed, Benzimidazole Derivatives: A Privileged Scaffold with Diverse Pharmacological Horizons, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 01-14 https://doi.org/10.5281/zenodo.17015431
 10.5281/zenodo.17015431
10.5281/zenodo.17015431