
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,2School of Pharmacy, P.P. Savani University, Kosamba, Surat, Gujarat-394125.
3Department of Pharmaceutical Chemistry, KLE College of Pharmacy, Vidyanagar, Hubballi-580031.
This review provides a comprehensive and up-to-date exploration of indole and its derivatives, focusing on their evolving role in medicinal chemistry. Indole, a privileged heterocyclic scaffold found in numerous natural and synthetic compounds, has demonstrated remarkable versatility in drug development due to its structural similarity to tryptophan and ability to interact with a wide range of biological targets. Through this review, readers will gain an in-depth understanding of the diverse pharmacological activities of indole-based compounds, including their anticancer, antimicrobial, anti-inflammatory, antioxidant, antidiabetic, and anti-leishmanial properties. The review also highlights key synthetic strategies, from classical methods such as Fischer and Madelung syntheses to innovative green and microwave-assisted approaches, showcasing how these techniques influence biological activity and drug-likeness. Detailed SAR analyses and molecular docking analysis are included to illustrate how specific substitutions and hybrid structures enhance potency and selectivity. Additionally, the review addresses emerging challenges in drug resistance and therapeutic failure, emphasizing how indole derivatives offer solutions through multitarget-directed ligand (MTDL) strategies. By reading this review, researchers and medicinal chemists will be equipped with critical insights into current trends, promising lead compounds, and future directions for developing indole-based therapeutics across multiple disease areas.
Because of its distinct structural and electrical characteristics, indole, a bicyclic aromatic heterocycle made up of a pyrrole moiety fused to a benzene ring, has become one of the most favored scaffolds in medicinal chemistry. It is a crucial framework in the identification and development of new therapeutic medicines due to its structural similarity to the important amino acid tryptophan and its occurrence in a wide variety of natural and synthesized bioactive molecules. Indole is found at the core of many pharmacologically significant molecules, including the neurotransmitter serotonin, the hormone melatonin, the anticancer drug vincristine, and the anti-inflammatory agent indomethacin.(1) Furthermore, approved drugs such as indomethacin (anti-inflammatory), Panobinostat (anticancer), and delavirdine (antiviral), along with endogenous biomolecules like serotonin and melatonin, underscore the broad biological relevance of this scaffold. Recent studies have highlighted the potential of indole derivatives in addressing multidrug resistance, modulating apoptotic and ferroptosis pathways, and inhibiting key enzymes such as CDK9, HDACs, PIM-1, and aromatase.(2) The indole nucleus is also known for its versatile pharmacological activities, including anticancer, antimicrobial, anti-inflammatory, antidiabetic, and neuroprotective effects. These compounds act on diverse molecular targets and signalling pathways, making them attractive candidates for multitargeted therapies. Since 2020, numerous novel indole-based compounds have demonstrated potent efficacy against cancer, bacterial, fungal, and viral pathogens, as well as metabolic disorders like diabetes and hypertension. Additionally, their ability to modulate inflammation, oxidative stress, and cellular signalling reinforces the ongoing significance of the indole scaffold in modern drug discovery and medicinal chemistry(3).
Fig.1
The objective is to equip researchers, particularly those in medicinal chemistry, pharmaceutical sciences, and chemical biology, with a solid foundation of knowledge that can accelerate the rational design of novel indole-based therapeutics. By elucidating SAR patterns and highlighting innovative synthetic strategies, the review intends to facilitate lead optimization, guide target selection, and support interdisciplinary approaches in drug discovery and development. The need for this study arises from several pressing global health challenges. The alarming increase in multidrug resistance, therapeutic failures, and the limited efficacy of existing drugs underscores the urgency to identify and develop new pharmacophores with improved activity and safety profiles. The burden of complex diseases such as cancer, tuberculosis, malaria, and chronic inflammatory disorders continues to rise, particularly in low- and middle-income countries, creating a critical demand for novel, multifunctional drug candidates. Indole, with its broad-spectrum biological activity and adaptability for chemical modification, represents a strategically significant scaffold for meeting this therapeutic need. Despite the availability of several studies on indole derivatives, much of the existing literature remains fragmented, with limited consolidation of recent findings. This review, therefore, serves to bridge this gap by integrating the latest research developments in one cohesive resource. It not only summarizes current achievements but also identifies emerging trends, challenges, and future directions, thereby laying the groundwork for continued innovation and potential clinical translation of indole-based therapeutics.
General synthetic methods for Indole:(4)(3)
Developed by Emil Fischer in 1883, the Fischer Indole Synthesis is still the most popular and researched technique for creating indole rings, which are essential building blocks in medicines and natural goods. The main focus of the reaction is the cyclization of arylhydrazones, which are created when aromatic hydrazines condense with carbonyl substances (such ketones or aldehydes). Arylhydrazoles may be made from a variety of starting materials, including diketones, ketones, aldehydes, keto acids, and keto esters. The arylhydrazone undergoes a sequence of ring closure steps and rearrangements that result in an indole derivative when heated and in the presence of an acidic catalyst, such as a Lewis acid (such as ZnCl?, FeCl?, or PCl?) or a protic acid (such as HCl, H?SO?, or TsOH).
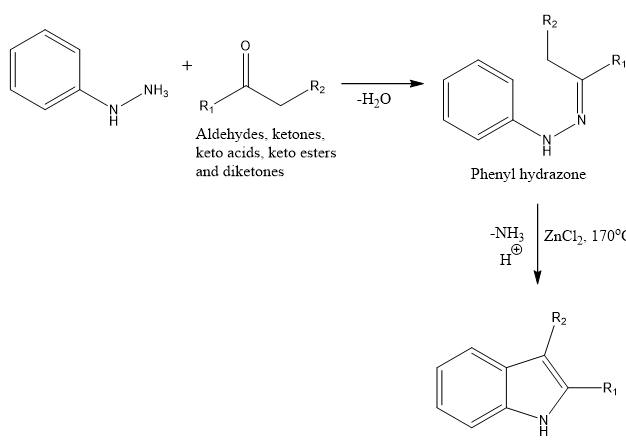
Scheme 1
Madelung synthesis:
The potential of ortho-alkyl-substituted anilides as precursors for the synthesis of conjugated indoles was originally identified by Madelung in 1912. These substrates are readily available, which is what makes this technique appealing.However, the harsh reaction conditions required for the process present certain drawbacks. These challenges have driven chemists to explore alternative approaches and refine the procedure over the years. Although this area has seen significant advancements, research has persisted into the past decade, leading to further enhancements and optimizations of the method. Under exceptionally harsh circumstances, the reaction entails the base-induced cyclization of 2-(acylamino)-toluene derivatives, usually requiring potassium tert-butoxide or sodium amide as the base at high temperatures between 250 and 300°C. Simple indole derivatives, such as 2-methylindoles, are best produced using this approach; compounds having sensitive functional groups cannot be produced using this method.
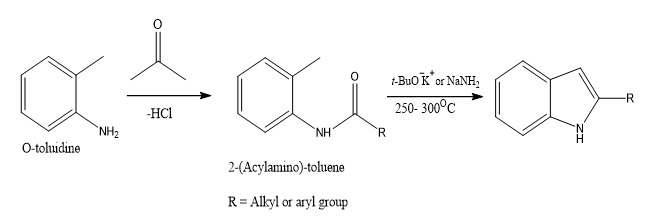
Scheme 2
2-arylamino-ketones, which are produced using a 2-halo-ketone and an arylamine, undergo acid-mediated cyclization as the reaction progresses, closing the aromatic system's electrophilic rings. Rearrangement events throughout the reaction often result in product mixes.
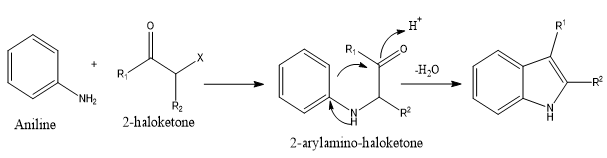
Scheme 3
Review of literatures on various activities of indole derivatives:
Bin Jia et, al., suggested a methodical strategy for the synthesis of diketopiperazine (DKP) alkaloids based on indole, focusing on three important analogues. 24 indole DKP derivatives were synthesized and evaluated for antibacterial activity as part of their inquiry, with an emphasis on comprehending their structure–activity correlations (SAR). Derivatives 3b and 3c of the produced compounds demonstrated strong antibacterial activity against all four tested bacterial strains. Additionally, components 4a and 4b had wide-ranging antimicrobial actions, showing effectiveness against a variety of plant pathogenic fungi in addition to their antibacterial qualities. Compound 3c's strong binding affinity for the FabH enzyme in Escherichia coli was shown by molecular docking investigations, providing information about its possible mode of action. All of these findings demonstrate the synthetic indole DKP compounds' therapeutic value and their potential as building blocks for the creation of new antibacterial drugs that specifically target FabH.(5).
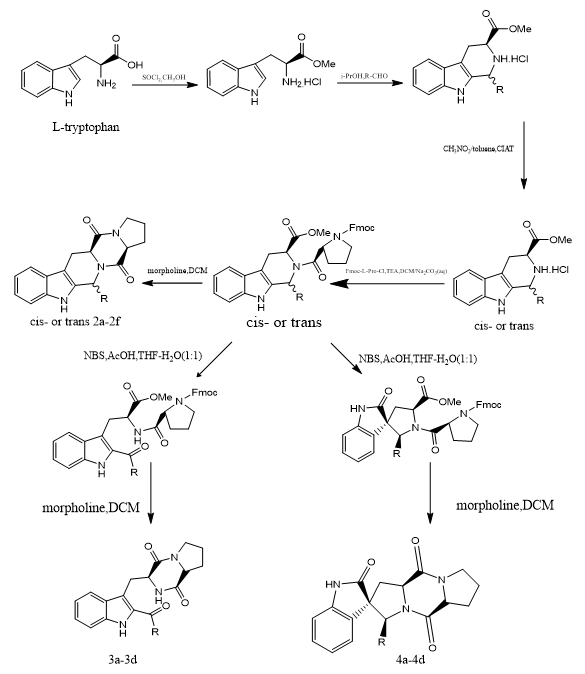
Scheme 3
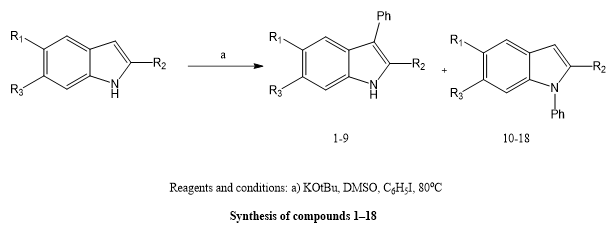
Tanveer MahamadAlli Shaikh et, al., highlighted how important indole is as a fundamental framework for creating new medicinal medicines. They used readily available starting materials to manufacture a number of N-substituted indole derivatives . Using the disc diffusion technique, the antibacterial activity of these substances was assessed in vitro against the albicans Candida species, Escherichia coli, Streptococcus and Staphylococcus aureus, respectively. Compound 1, also known as 4-(1-(2-(1H-indol-1-yl)ethoxy)pentyl)-N,N-dimethylaniline, was the most effective antibacterial of the produced molecules, outperforming both its structural equivalents (compounds 2 and 3) and the common antibiotic chloramphenicol. This suggests that compound 1 has significant potential as a lead chemical in the development of effective antibacterial drugs(6).
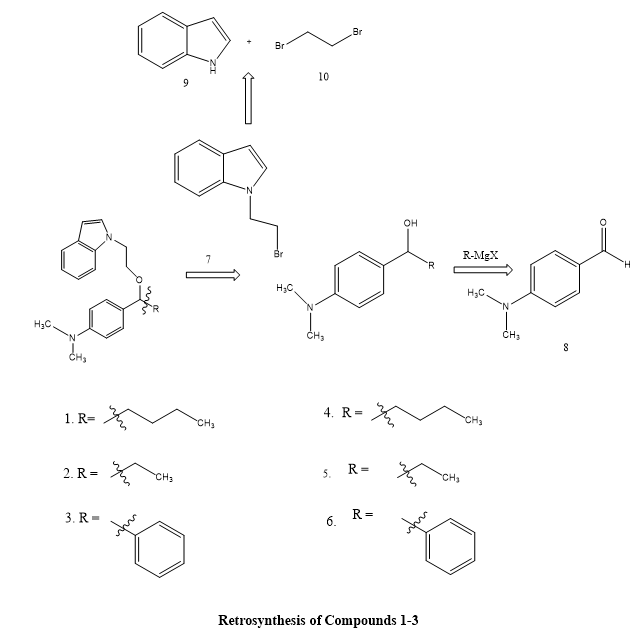
Scheme 4
Khadhar Navaz Umar Basha et, al., described an effective and simple one-pot synthesis technique for generating 3-(1H-indole-3-carbonyl)-2H-chromen-2-one substrates. Through the use of a water-methanol (1:1) solvent solution, 3-cyanoacetylindole and salicylaldehyde were reacted in the presence of sodium carbonate. The methodology has various advantages, including broad substrate compatibility, high product yields, simplicity of operation, and easy purification processes, making it a viable method for synthesizing indole-chromenone hybrids. The study also included the first assessment of their in vitro antioxidant and antibacterial properties. Strong scavenging of free radicals over DPPH was demonstrated by molecules 5e, 7a, and 7b. Additionally, substances 5f, 7b, and 5g showed higher reducing power, with EC50 values lower than those of the conventional antioxidants ascorbic acid (AA) and butylated hydroxyanisole (BHA). Antibacterial testing found that compounds 5d and 5f were efficient against methicillin-resistant Staphylococcus aureus (MRSA), whereas compounds 5c, 7a, and 7b were active against Bacillus species. Compounds 5b and 5e exhibited significant activity over a Gram-negative bacteria while substances 5h and 5g shown encouraging antifungal efficacy, namely against Candida albicans (7).
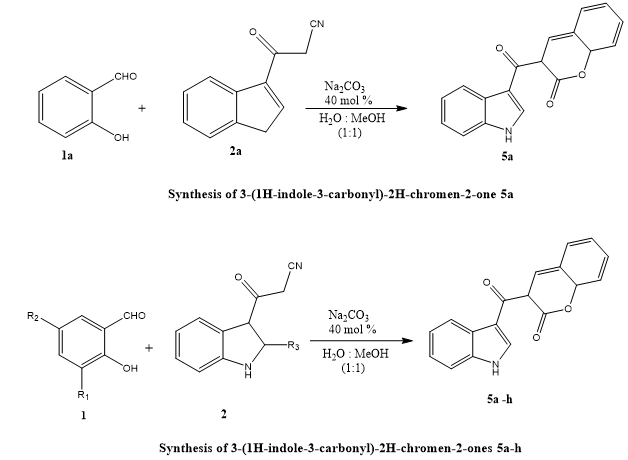
Scheme 5
Khushbu Agrawal et, al., highlighted the growing significance of indole and its derivatives in a range of biological activities and emphasized its value in medicinal chemistry. Antitubercular, antiviral, antibacterial, and antimalarial, anti-inflammatory in nature antileishmanial, anticholinesterase, anticancer, and enzyme inhibitory qualities are a few these comprise. The WHO has identified antimicrobial resistance (AMR) as one of the top ten global health threats due to its increasing prevalence. New therapeutic medications are desperately needed. In this study, we created novel indole derivatives based on sulphonamides from 1H-indole-2-carboxylic acid. 1H NMR and LC-MS spectroscopy were used to validate the structural identity of the produced compounds. Both Gram-positive and Gram-negative bacterial strains were used to assess the antibacterial activity of these compounds. Among the investigated substances, good activity was demonstrated against Staphylococcus aureus, with particularly high inhibitory effects against Klebsiella pneumoniae, showing potential as effective antibacterial agents(8).
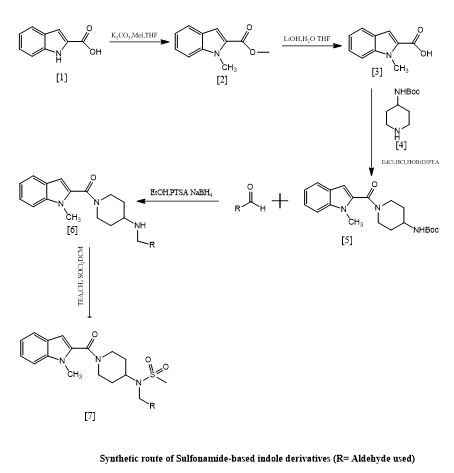
Scheme 6
Reem I. Al-Wabli et, al., highlighted the growing worry about microbial infections and the increasing resistance to conventional antimicrobial medicines, highlighting the critical need for new compounds with superior pharmacological characteristics. In response, a novel series of conjugates of indole and triazole were developed and produced as possible antibacterial agents. These compounds antibacterial activity was assessed in vitro utilizing inhibition zone diameter (DIZ) and MIC testing against certain strains of microorganisms. With inhibition zones measuring 11 to 15 mm and MIC values of around 250 µg/mL, the study discovered that several compounds had moderate to outstanding antibacterial effectiveness against Gram-negative bacteria. Excellent results were obtained from the antifungal evaluation, particularly against Candida tropicalis. Having concentrations of the MIC lower than as 2 µg/mL, a number of drugs demonstrated notable activity. With the minimum inhibitory concentration of 2 µg/mL, compound 6f showed the best antifungal effectiveness against Candida albicans, suggesting that it might be used as a lead molecule for the development of antifungal drugs.(9).
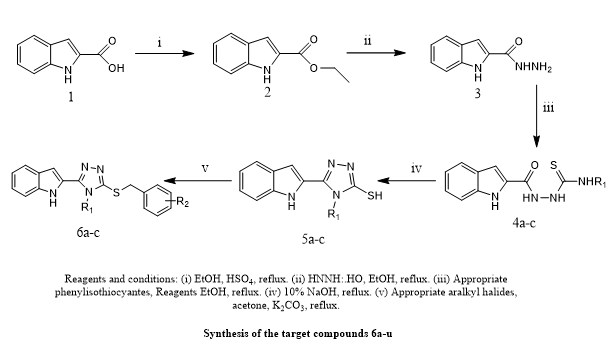
Scheme 7
Aboubakr H. Abdelmonsef et, al., designed and incorporated five- and six-membered heterocycles, such as pyrazole, furo[2,3-b], indole, and chromene moieties, into three new series of quinazolin 2,4-dione-based acylthiourea derivatives. (compound 2) was produced in a one-pot, three-component reaction combining a benzoyl chloride derivative (compound 1), cyanoacetic acid hydrazide and ammonium thiocyanate. Knoevenagel condensation of compound 2 with different aromatic and heteroaromatic aldehydes or ketones resulted in the synthesis of the final quinazolinone-arylthiourea conjugates in high yields (60-75%). At low concentrations in vitro, 3b and 3c, two of the synthesized compounds, showed strong inhibitory effects against a number of bacterial and fungal strains, indicating noteworthy antimicrobial activity. The chemicals' binding interactions with important microbial enzymes, including Candida albicans dihydrofolate reductase (DHFR), Pseudomonas aeruginosa lipase, and Staphylococcus aureus tyrosyl-tRNA synthetase, were assessed using molecular docking experiments. The benchmark medications for the antibacterial and antifungal comparisons were nystatin and ciprofloxacin, respectively. Studies of the structure–activity relationship (SAR) showed that particular functional groups helped compounds 3b and 3c become more potent. Furthermore, the geometric optimization of these promising drug candidates was facilitated by semiempirical modeling.. According to these results, compounds 3b and 3c have a great deal of promise as an lead candidates for the creation of innovative antibacterial medications (10).

Scheme 8
Marvin Fresia et, al., devised an innovative synthetic strategy utilizing a photo-Nazarov cyclization of a 3-acylindole precursor, initially leading to the formation of a thermodynamically stable cis-hydrindanone. This intermediate underwent reduction and was transformed into a cyclopentadiene, followed by dihydroxylation and hydrogenation steps to obtain the corresponding indoline. A key feature of the synthesis was a stereospecific hydride migration within a dioxaphospholane intermediate under Grainger's conditions—marking the first successful application of this method to an N-heterocyclic system. Starting from an indole scaffold, the researchers were able to generate previously unreported methanocyclohepta [b]indolone derivatives. To achieve deoxygenation of the trans-hydrindanone, the compound was first converted into a 1,3-dithiolane and then reduced using RANEY® Ni. The resulting trans-hydrindanone was deprotected with sodium naphthalide and further dehydrogenated using palladium on charcoal, ultimately yielding the desired trans-fused indeno[1,2-b]indole (compound 24). This product was obtained in a 2.5% overall yield across a 12-step synthetic route. Overall, the study illustrates the potential of the photo-Nazarov approach in assembling intricate indole-fused systems while effectively addressing stereochemical challenges(11).
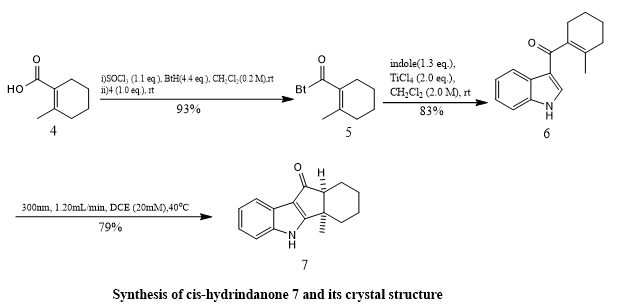
Scheme 9
Syeda Laila Rubab et, al., the researchers conducted an in-depth study aimed at synthesizing and analyzing bis(4,6-dimethoxy-2,3-diphenyl-1H-7,7′-indolyl)methane, along with a few structurally similar indole-based compounds. Their synthetic pathway began with a modified version of the Bischler indole synthesis to produce an initial compound (compound 1). This compound underwent a formylation reaction to yield compound 2, which was then chemically reduced to form an alcohol intermediate (compound 3). When efforts were made to cyclize this alcohol into a fused heterocyclic structure (intended compound 5), the reaction unexpectedly gave rise to a different product—a bisindole compound identified as compound 4. The structure of this compound was thoroughly confirmed using advanced spectroscopic tools and single-crystal X-ray diffraction, ensuring accuracy in its molecular identification. Revealing a non-planar geometry with distinct dihedral angles. Hirshfeld surface analysis was employed to evaluate intermolecular interactions, showing that hydrogen-hydrogen and hydrogen-carbon contacts dominated, while enrichment ratio calculations confirmed that hydrogen-oxygen interactions played a crucial role in crystal packing. Density Functional Theory (DFT) studies provided insights into the electronic properties, including HOMO–LUMO energy gaps, chemical potential, electronegativity, and electrophilicity. Among the synthesized compounds (1–4), the bisindole derivative (4) exhibited the highest stability and most favorable electronic properties, suggesting its potential as a strong pharmacological candidate. Furthermore, both in vitro and in silico evaluations of antibacterial, antiurease, and α-amylase inhibitory activities revealed notable bioactivity, particularly for the dimerized indole, underscoring the therapeutic promise of these indole-based compounds(12).
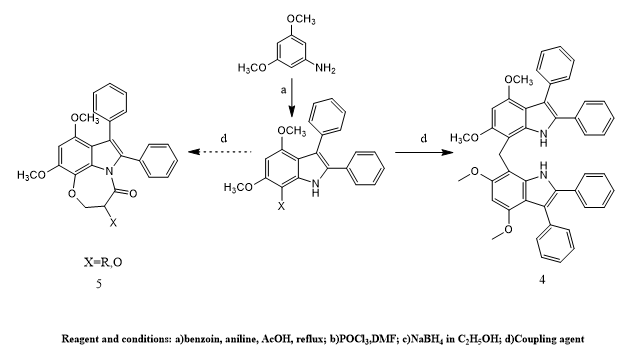
Scheme 10
Mona F. Said et, al., Researchers aimed to develop multi-target directed ligands as a therapeutic strategy to address the multifaceted nature of Alzheimer’s disease. They designed a novel series of 3-hydrazinyl indole phenacetamide derivatives intended to inhibit three key enzymes implicated in AD progression— BChE ,AChE, and β-secretase each of which contributes to neurodegeneration and inflammation. Initial in vivo anti-inflammatory screening identified compounds 5a–f, 5h, 5j, and 5o as promising leads, which were then subjected to in vitro AChE inhibition assays. Among them, compounds 5a–c, 5j, and 5o displayed strong inhibitory activity against both AChE and BChE and were further evaluated in animal models. Additionally, compounds 5a–c was analyzed for their impact on hallmark AD features and brain histological changes. Notably, compound 5a—an N-phenylacetamide indole derivative with an unsubstituted phenylhydrazinyl group—demonstrated superior cognitive benefits compared to the standard drug donepezil. It enhanced spatial memory, reduced histopathological damage, and decreased levels of major AD biomarkers such AChE, BACE1, Aβ, and p-Tau. It also improved oxidative stress (GSH) and the inflammatory response (IL-1β).In vitro, 5a-c exhibited moderate BACE1 inhibition. In silico molecular docking revealed that these derivatives have strong binding affinities for key residues in the AChE and BACE1 active sites. Molecular dynamics simulations confirmed compound 5a's structural stability within both enzyme targets over the simulation timeframe. Taken together, compound 5a stands out as a promising multi-target lead molecule for the treatment of AD.combining efficient inhibition of AChE, BChE, and BACE1 with neuroprotective and anti-inflammatory effects(13).
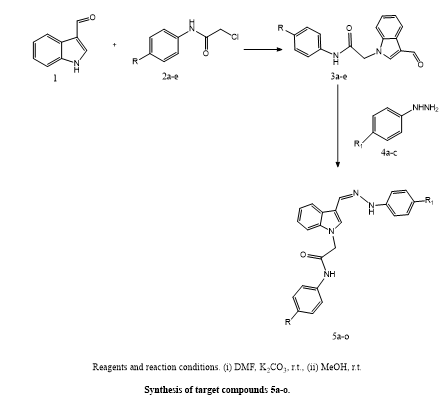
Scheme 11
Chao Zhang et, al., developed a distinct class of indole-containing benzamide compounds aimed at targeting two essential functions of the influenza virus RNA-dependent RNA polymerase complex. These molecules were specifically engineered to interfere with two critical viral processes: the interaction at the PA subunit’s C-terminal domain and the oligomerization of the viral nucleoprotein (NP), both vital for efficient viral replication. Among the newly synthesized compounds, 8e and 8f emerged as the most effective, showing strong antiviral effects with EC?? values of 1.64 ± 0.05 μM and 1.41 ± 0.27 μM, respectively, against the H1N1 strain, and exhibited broad activity against multiple influenza A and B strains. Unlike older benzofurazan analogs, these candidates displayed low cytotoxicity, with CC?? values exceeding 100 μM, indicating a favorable safety margin. Binding studies using surface plasmon resonance (SPR) revealed high affinity for both the PAC domain and NP, supporting their dual-target mechanism. A range of computational techniques—including molecular docking, molecular dynamics simulations, dynamic correlation analysis, principal component analysis (PCA), and quantum-level DFT calculations—were employed to investigate how these compounds interact at the molecular level and what structural features contribute to their potency. This study not only offers promising leads for the development of anti-influenza agents but also showcases an effective molecular design strategy for creating safe and multifunctional antiviral compounds targeting essential viral machinery(14).
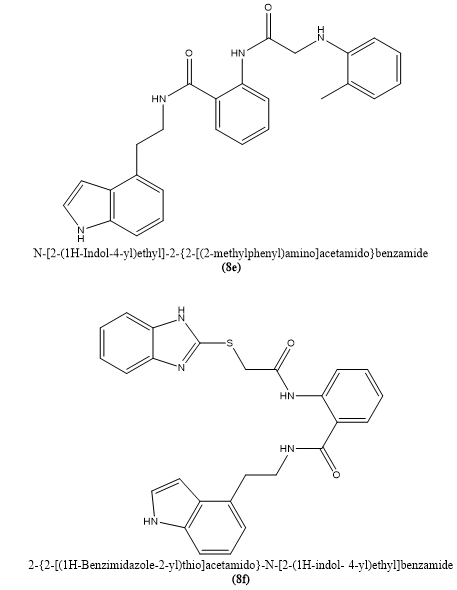
Fig. 2
Andrea Citarella et, al., created a A new generation of ethyl cinnamate compounds incorporating an indole core structure has been designed as potential broad-spectrum antiviral agents with activity against coronaviruses. What sets these compounds apart is their ability to simultaneously engage both viral proteins and host cell enzymes involved in the infection cycle. Specifically, they were optimized to inhibit two key viral and host targets: the SARS-CoV-2 main protease (Mpro), which is essential for processing viral polyproteins, and the host proteases cathepsin L (CatL) and cathepsin S (CatS), which facilitate viral entry into human cells. Enzyme-based assays confirmed that the newly synthesized compounds effectively suppressed the activity of these targets. Further investigation into structure–activity relationships (SAR) pinpointed the tert-leucine (Tle) residue at the P2 site of the molecular scaffold as a critical factor contributing to the dual inhibition capability. Among the series, compounds labeled 12, 20, and 3 were particularly noteworthy, exhibiting antiviral activity at low micromolar concentrations against both alpha-coronavirus (hCoV-229E) and beta-coronavirus (hCoV-OC43). Their antiviral efficacy was accompanied by favorable selectivity indices—98 for compound 12, 56 for compound 20, and 101 for compound 3—indicating strong antiviral action with minimal toxicity. Mechanistic studies revealed that compound 12 acts primarily at the stage of viral entry by targeting CatL, effectively disrupting the host-mediated step of infection. In contrast, compound 20 displayed a broader mode of action by concurrently targeting CatL and Mpro, thereby interfering with both early and late stages of the viral life cycle. These multitarget strategies enhance antiviral potency and minimize the likelihood of resistance development. Overall, the research provides strong evidence that indole-based cinnamate derivatives containing the Tle unit represent a robust chemical platform for the development of antiviral drugs. Their ability to act on multiple biological pathways involved in coronavirus infection supports their potential utility not only in current therapeutic efforts but also as a strategic asset in addressing future pandemics.(15).
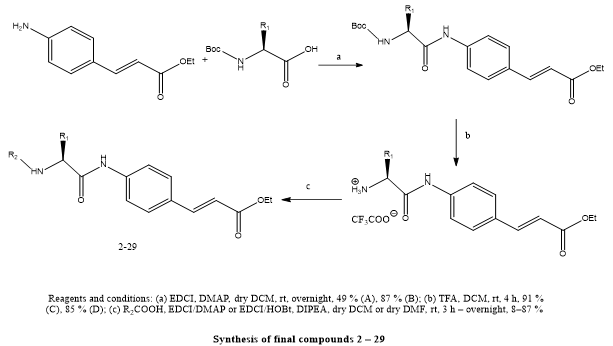
Scheme 12
Lu-yao Zhao et, al., addressed the pressing need for innovative treatments in hepatocellular carcinoma (HCC), which has a five-year survival rate of only 20%. The study focused on inhibiting ULK1, a key autophagy-promoting kinase, as a potential therapeutic strategy. Virtual docking simulations were employed to predict how XST-14 derivatives interact with the ULK1 protein, which guided the rational design and subsequent synthesis of four new chemical entities. Among these, derivative A4 stood out as a promising contender, with tenfold more activity than XST-14 and superior anti-HCC activities than the conventional medicine sorafenib. A4 effectively inhibited tumor development and improved the therapeutic effectiveness of sorafenib in HCCLM3 & HepG2 cell lines. Notably, A4 proved effective against sorafenib-resistant HCC in both vitro and vivo. These findings back up ULK1 inhibition as a promising strategy to HCC therapy and highlight A4 as a possible lead chemical with acceptable safety and effectiveness characteristics, providing fresh hope for improving HCC patient outcomes(16).
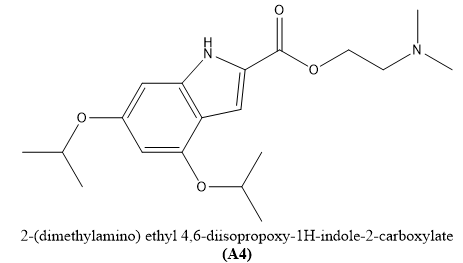
Fig.3
Shoaib Khan et, al., created and A novel series of sulfonothioate derivatives containing both indole and triazole moieties (compounds 1–15) was synthesized with the goal of identifying potential antidiabetic agents. The chemical structures of the synthesized compounds were thoroughly characterized using advanced analytical techniques, including proton (^1H) and carbon (^13C) nuclear magnetic resonance (NMR) spectroscopy, as well as high-resolution electron ionization mass spectrometry (HREI-MS). The antidiabetic potential of these compounds was evaluated in vitro, with their inhibitory effects compared against the standard reference drug, Acarbose. Among the tested derivatives, compounds 7, 9, and 15 demonstrated superior inhibitory activity, recording lower IC?? values than Acarbose. Specifically, compound 7 exhibited IC?? values of 3.20 ± 0.10 µM and 4.20 ± 0.50 µM; compound 9 showed values of 5.10 ± 0.10 µM and 5.70 ± 0.30 µM; and compound 15 displayed IC?? values of 3.20 ± 0.70 µM and 3.40 ± 0.50 µM, suggesting stronger potency. Structure–activity relationship (SAR) analysis highlighted that both the nature and position of substituents—such as fluorine, chlorine, hydroxyl, and nitro groups—played a pivotal role in modulating biological activity. Additionally, molecular docking simulations demonstrated favorable binding interactions between the most active compounds and their target enzymes, further validating their potential as enzyme inhibitors. Pharmacokinetic profiling through ADME (absorption, distribution, metabolism, and excretion) studies indicated that these lead compounds possessed drug-like properties aligning with accepted pharmaceutical benchmarks, supporting their suitability for further development as antidiabetic therapeutics(17).
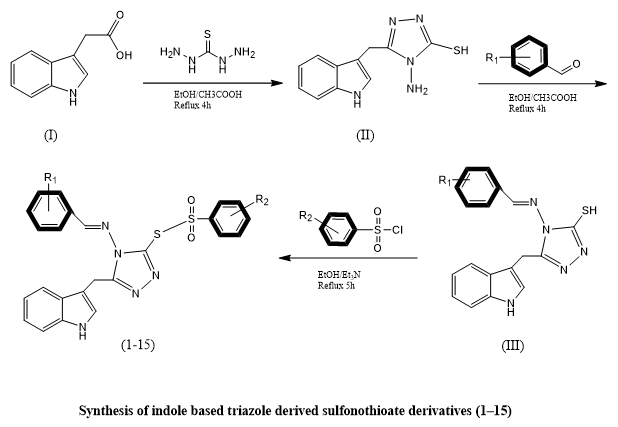
Scheme 13
De-Cai Dai et, al., isolated three previously unreported indole-based alkaloids, named fissindoalkas A–C (compounds 1–3), along with one previously identified structurally related indole derivative (compound 4), were extracted from the roots of Fissistigma oldhamii (Hemsl.) Merr. The chemical identities and structures of these compounds were elucidated through comprehensive spectroscopic techniques. Their anti-inflammatory potential was assessed based on their ability to inhibit nitric oxide (NO) production in RAW264.7 murine macrophage cells stimulated with lipopolysaccharide (LPS). Among the isolated compounds, derivatives 2 and 3 exhibited notable inhibitory activity, with IC?? values of 2.52 ± 0.18 µM and 2.33 ± 0.16 µM, respectively. These results highlight the potential of indole alkaloids from F. oldhamii as promising natural candidates for developing novel anti-inflammatory therapeutics.(18).
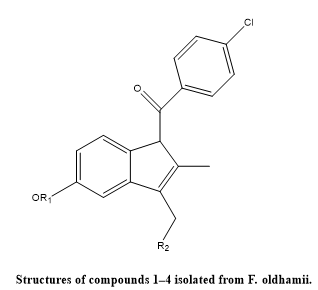
Fig.4
Yandan Wu et, al., in response to the increasing incidence of Candida albicans infections that no longer respond to conventional antifungal treatments, a novel group of indole-derived compounds was synthesized and investigated for their therapeutic potential. The antifungal efficacy of these molecules was evaluated under in vitro conditions, where several candidates demonstrated notable inhibitory effects against fluconazole-resistant C. albicans strains. Notably, their activity was evident both as individual agents and in combination with fluconazole, indicating a potential for synergistic action and enhanced treatment outcomes. Further analysis through structure–activity relationship studies allowed researchers to pinpoint specific structural elements within the indole scaffold that were crucial for antifungal effectiveness. Modifications to ring substituents, side chains, and electron-donating or -withdrawing groups were shown to directly influence the compounds' biological performance. These findings provide valuable guidance for future molecular optimization aimed at enhancing potency, selectivity, and overcoming resistance mechanisms in fungal pathogens. Compound 1 stood out among the produced compounds because it efficiently inhibited key pathogenic pathways such as yeast-to-hypha transformation and biofilm development. Mechanistic studies revealed that compound 1 increased efflux pump activity, disrupted mitochondrial function, and decreased intracellular ATP levels, indicating a multi-targeted mechanism of action. In vivo investigations indicated its therapeutic potential, with strong antifungal effects shown on histological inspection. Overall, compound 1 shows great promise as a novel antifungal agent for combating drug resistance in Candida albicans(19).
Scheme 14
Jianzhang Wu et, al., created a Researchers introduced a novel class of spirocyclic compounds derived from the fusion of 1H-benzo[e]indole-2(3H)-one and 1,4-dihydroquinoline frameworks, aiming to harness pyroptosis—a form of programmed cell death—as a strategy for cancer treatment. These hybrid molecules were synthesized through a sustainable and environmentally conscious one-pot reaction, using 10 wt% sodium dodecyl sulfate (SDS) in water, representing a green, atom-economical approach with minimal environmental impact. The biological activity of the synthesized compounds was assessed via MTT assays to evaluate their anti-tumor properties. Several derivatives showed considerable inhibitory effects on cancer cell growth. Further computational analysis using quantitative structure–activity relationship (QSAR) modeling, built with a random forest algorithm (R² = 0.9656 and 0.9747), highlighted compound A9 as the most promising candidate. In vitro assays confirmed that A9 suppressed ovarian cancer cell proliferation, migration, and invasion in a concentration-dependent manner. Mechanistic investigations revealed that A9 induced pyroptotic cell death by upregulating GSDME-N, a key effector protein in pyroptosis, while simultaneously promoting apoptosis by modulating the expression of apoptosis-regulating proteins. In vivo studies reinforced these findings, where treatment with A9 at a dosage of 5 mg/kg led to a marked reduction in tumor volume and mass. This was accompanied by increased GSDME-N expression and decreased caspase-3 levels in tumor tissues. Collectively, this study demonstrates a resource-efficient synthesis method and positions compound A9 as a potent pyroptosis-inducing agent with robust anticancer efficacy, making it a strong candidate for future therapeutic development(20).
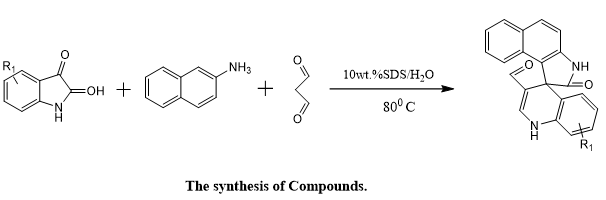
Scheme 15
Anjela Xalxo et, al., created a novel 3-sulfenylindole derivatives was synthesized through an oxidative cross-dehydrogenative coupling reaction involving 4-hydroxy-2H-chromene-2-thione and indole. This transformation was achieved under mild and environmentally considerate reaction conditions, utilizing 10 mol% molecular iodine as the catalyst and tert-butyl hydroperoxide (TBHP) as the oxidant in dimethyl sulfoxide (DMSO) at ambient temperature. The method demonstrated high efficiency and aligns with green chemistry principles, making it a sustainable approach for constructing biologically relevant scaffolds. Given their favorable physicochemical profiles and potential pharmaceutical relevance, selected members of this compound series—namely 3a, 3b, 3d, 3f, 3h, and 3k—underwent further oxidative modification to yield their corresponding sulfone derivatives. These structural transformations not only expand the chemical diversity of the synthesized library but also contribute to exploring their potential as future drug candidates by enhancing their metabolic stability and bioactivity. Molecular docking and target prediction studies were performed on six sulfone derivatives (5a-5f), four of which (5a, 5d, 5e, and 5f) demonstrated significant binding affinity and were chosen for in vitro evaluation. These sulfones showed strong anti-proliferative activity against MCF7 breast cancer cells. Additionally, quantum chemistry research found that the reaction is exergonic, supporting its thermodynamic viability. Overall, this study describes a flexible and sustainable approach for producing physiologically active sulfenylindole sulfones with potential anticancer uses(21).
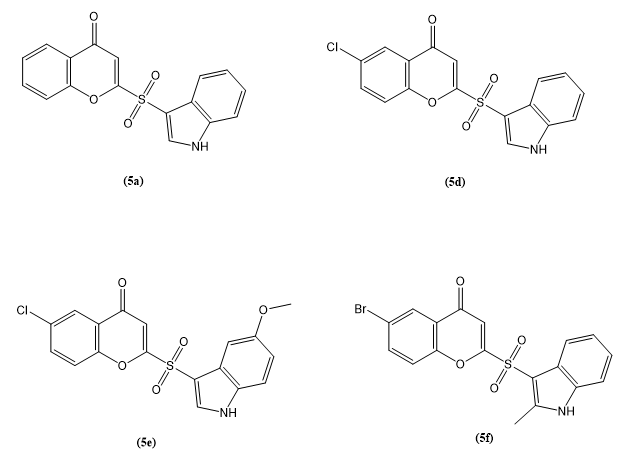
Fig.5
Eldehna et, al., worked on the creation of new PIM-1 kinase inhibitors, which are a potential technique in cancer therapy due to PIM-1's critical involvement in cell proliferation, survival, and resistance to therapy. This study focused on the synthesis and biological evaluation of two distinct compound classes—pyrazolo[1,5-a]pyrimidines (6a–i) & pyrazolo[3,4-b]pyridines (10a–i)—for their potential anticancer effects. The cytotoxicity of these molecules was assessed across a panel of human cancer cell lines, including A-549 (lung carcinoma), PANC-1 (pancreatic carcinoma), and A-431 (epidermoid carcinoma), along with normal lung fibroblast cells (MRC5) to evaluate selectivity and safety. Among the tested compounds, 6b, 6c, 6g, 6h, 6i, and 10f exhibited significant antiproliferative activity against A-549 cells, showing IC?? values in the range of 1.28 to 3.52 μM, while demonstrating minimal cytotoxicity toward normal cells. Of particular interest was compound 10f, which features a 4-methoxyphenyl substituent and displayed potent inhibition of PIM-1 kinase, with an IC?? of just 0.18 μM. Further in vitro studies revealed that 10f induced programmed cell death (apoptosis) and caused cell cycle arrest in A-549 cells, pointing to its mechanism of action. Computational studies, including molecular docking and molecular dynamics simulations, supported these findings by showing stable and high-affinity interactions between the compound and the active site of PIM-1 kinase. Overall, compound 10f stands out as a promising and selective inhibitor of PIM-1, with strong potential for development as a targeted anticancer agent. (22)
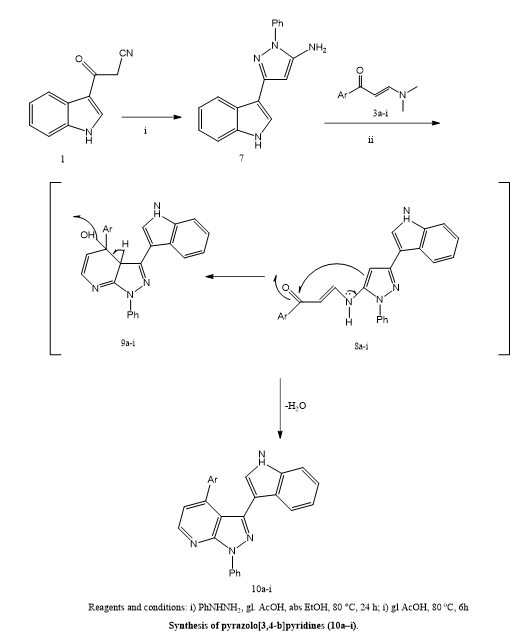
Scheme 16
Phoebe F. et, al., created and synthesized fourteen new indole-pyrimidine hybrid compounds with the goal of targeting the anti-apoptotic protein Mcl-1, which is widely overexpressed in many malignancies. The synthesized compounds' structures were verified using spectroscopic methods. The (E)-configuration of the synthesized compounds was confirmed through a combination of computational and experimental methods, utilizing MM2 force field calculations alongside 2D NOESY NMR spectroscopy for structural validation. Initial biological evaluations indicated that several compounds—particularly 7d, 7e, 7i, and 7k—exhibited potent inhibitory activity against the anti-apoptotic protein Mcl-1, with Ki values falling within a narrow range of 11.19 to 15.21 nM. Additional profiling revealed that select molecules, such as 7i and 7d, also demonstrated dual inhibitory effects on Bcl-XL and Bcl-2 proteins, suggesting broader activity across the Bcl-2 family of apoptosis regulators. To further assess their anticancer potential, the most active compounds were subjected to in vitro cytotoxicity testing against a panel of human cancer cell lines, including PC-3 (prostate cancer), K-562 (chronic myelogenous leukemia), and MDA-MB-231 (triple-negative breast cancer). The results showed moderate to strong antiproliferative effects across these models. Computational docking studies provided further support by confirming stable binding interactions of these inhibitors within the Mcl-1 binding pocket. Additionally, in silico predictions of pharmacokinetic properties, including drug-likeness and pre-ADMET parameters, suggested that the compounds possess favorable profiles for further development as anticancer drug candidates.(23).
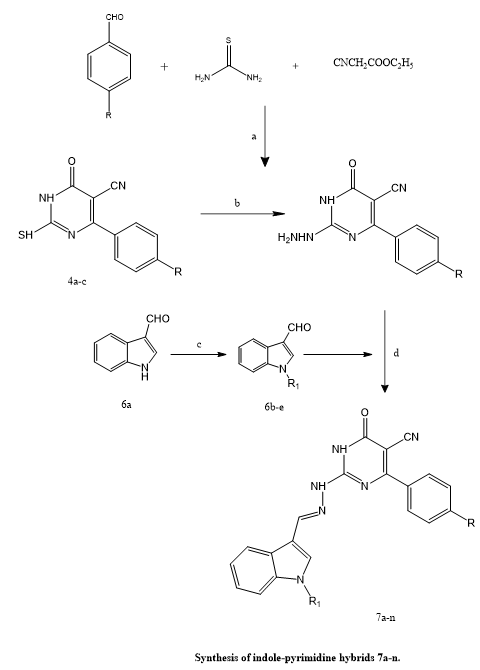
Scheme 17
Tianpeng Chen et, al., to address the urgent need for effective therapeutic options for acute lung injury (ALI) and acute respiratory distress syndrome (ARDS)—both of which are characterized by high mortality rates and limited treatment choices—a new class of 4-indolyl-2-arylaminopyrimidine derivatives was synthesized. A total of eleven compounds were developed and evaluated for their potential anti-ALI effects. Among them, compounds 6c and 6h emerged as the most promising, exhibiting strong anti-inflammatory properties by significantly reducing the levels of key pro-inflammatory cytokines interleukin-6 (IL-6) and interleukin-8 (IL-8) by approximately 62–77% and 65–72%, respectively. These effects were observed in vitro with minimal cytotoxicity, indicating favorable safety profiles. Further in vivo assessment using a mouse model of ALI demonstrated that compound 6h, administered at a dose of 20 mg/kg, markedly reduced inflammatory cell infiltration within lung tissues. This resulted in improved lung architecture and alleviated pathological damage. Mechanistic studies revealed that the therapeutic action of 6h is mediated through the suppression of p38 and ERK phosphorylation, key components of the mitogen-activated protein kinase (MAPK) pathway. By modulating this signaling cascade, the compound effectively dampens the inflammatory response. Collectively, these findings support compound 6h as a compelling lead candidate for the development of novel anti-inflammatory agents aimed at treating ALI. Its demonstrated efficacy in both cell-based and animal models highlights its potential for further preclinical and clinical investigation(24).
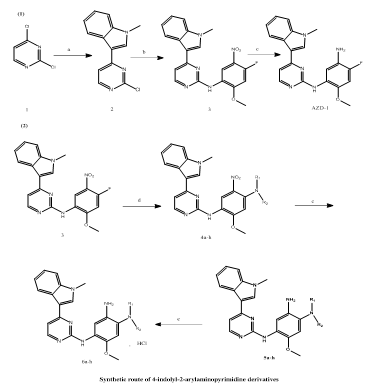
Scheme 18
Garvita Mishra et, al., in response to the urgent demand for new therapeutic agents to treat leishmaniasis, researchers developed and synthesized a collection of thirty-four indole–dihydropyrimidinone hybrid molecules using the Biginelli multicomponent reaction—a well-known, efficient synthetic approach. These compounds were evaluated for their anti-leishmanial activity through both in vitro and in vivo models. In vitro assessments were performed against intracellular amastigote forms of Leishmania donovani, while in vivo efficacy was determined using a golden hamster model representative of visceral leishmaniasis (VL). Among the synthesized compounds, derivatives 4f and 4m demonstrated notable inhibitory effects on the parasite, exhibiting IC?? values of 4.54 μM and 5.05 μM, respectively. Importantly, both compounds displayed minimal cytotoxicity toward the J774.1 murine macrophage cell line, indicating a favorable therapeutic index. These findings suggest that the indole–dihydropyrimidinone scaffold holds promise as a basis for developing effective and safe anti-leishmanial drug candidates. Notably, compound 4f demonstrated >65% parasite removal in vivo, greatly outperforming 4m. Mechanistic studies revealed that 4f acts by inducing oxidative stress, which causes mitochondrial dysfunction, ATP depletion, and, ultimately, apoptosis in the parasite. Additionally, SAR (structure-activity relationship) and pharmacokinetic (PK) investigations confirmed 4f's promise as a lead molecule for future anti-leishmanial medication development. In conclusion, compound 4f is a promising candidate for continued development as a chemotherapeutic agent for visceral leishmaniasis, addressing the crucial dilemma of drug resistance and limited treatment alternatives (25).
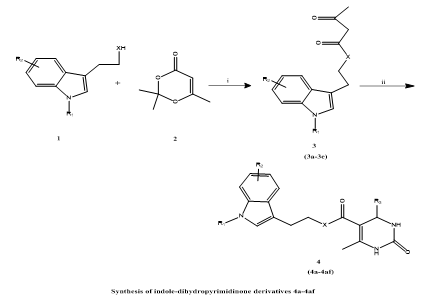
Scheme 19
Xiaohui Chen et, al., A novel series of N-(2-amino-phenyl)-5-(4-aryl-pyrimidin-2-ylamino)-1H-indole-2-carboxamide derivatives was designed and synthesized to act as dual inhibitors of cyclin-dependent kinase 9 (CDK9) and class I histone deacetylases (HDACs)—two critical enzymes involved in transcriptional dysregulation often linked to cancer progression. Within this series, compound 13ea emerged as a standout, exhibiting potent anti-proliferative effects with IC?? values below 5.0 μM across multiple cancer cell lines, including HeLa, MDA-MB-231, and HepG2. Biochemical assays demonstrated that 13ea effectively inhibited CDK9 phosphorylation (IC?? = 0.17 μM) and also suppressed the deacetylase activity of HDAC1 and HDAC3 with IC?? values of 1.73 μM and 1.11 μM, respectively. Molecular docking studies confirmed strong binding interactions between 13ea and the catalytic sites of CDK9, HDAC1, and HDAC3, supporting its dual-target mechanism of action. Cellular assays further showed that the compound induced G2/M phase cell cycle arrest and activated mitochondrial apoptotic pathways. When compared to existing drugs—AZD-5438 (a selective CDK9 inhibitor) and Mocetinostat (a class I HDAC inhibitor)—compound 13ea demonstrated superior efficacy. In vivo experiments using an MDA-MB-231 xenograft mouse model revealed that treatment with 13ea at a dose of 30 mg/kg led to a substantial tumor volume reduction of 76.83%. These results suggest that 13ea is a highly promising dual-acting anticancer agent with considerable therapeutic potential, warranting further preclinical development(26).
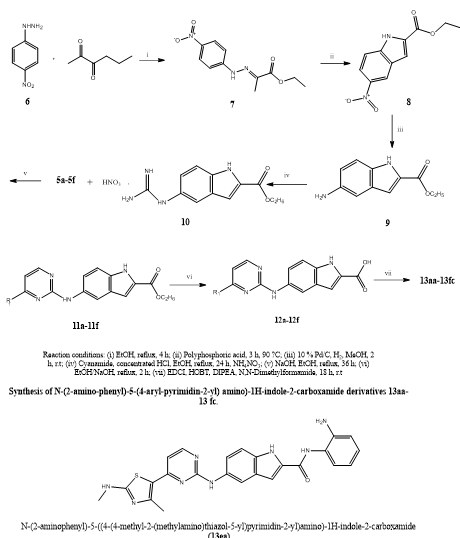
Scheme 20
Satya Kumar et, al., A novel series of thirty-two 1H-indole-based Meldrum-adjoined 1H-1,2,3-triazole derivatives was synthesized and evaluated for their potential as dihydrofolate reductase (DHFR) inhibitors. This unique set of compounds (2–13, 15a–15f, 16a–16f, 17a–17f, 19a, 19b, and 20a) represents a new structural class of triazole-linked heterocycles. The derivatives labeled 15a–15f, 16a–16f, and 17a–17f were constructed via copper(I)-catalyzed azide-alkyne cycloaddition ("click" chemistry), involving substituted indole-based Meldrum alkynes (compounds 11, 12, and 13) and aromatic azides (14a–14f) in the presence of CuI and Hünig’s base. Further chemical modifications of these intermediates led to compounds 19 and 20, through the selective cleavage of the Meldrum acid moiety. The molecular structures of key intermediates—specifically compounds 6, 11, and 13—were unambiguously confirmed by single-crystal X-ray diffraction analysis. The full library was subjected to in vitro DHFR inhibition assays. Many of the synthesized molecules exhibited notable inhibitory activity, with IC?? values ranging from 3.48 ± 0.16 to 30.37 ± 1.20 μM. Among them, ten compounds—namely 15c–15f, 16c–16f, 17e, and 17f—stood out with particularly strong DHFR inhibition, while the remaining derivatives showed moderate efficacy. To further understand their binding interactions, molecular docking studies were conducted. Compound 16f displayed the most favorable docking score (−9.13 kcal/mol), suggesting high binding affinity with the DHFR active site, and outperforming compound 15c (moderate activity) and compound 16a (weaker activity). These computational results were in line with the observed biological activity data. Overall, this work introduces a new and promising class of DHFR inhibitors based on Meldrum–triazole–indole hybrids, which may serve as valuable scaffolds for further development in medicinal chemistry and antifolate drug discovery.(27).
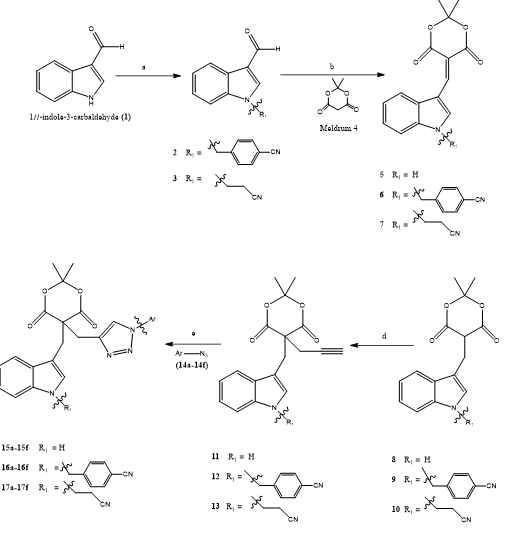
Scheme 21
Neha Bhatia et, al., investigates the potential of indole-based molecules as effective aromatase inhibitors for managing estrogen receptor-positive (ER?) breast cancer, which accounts for more than 70% of diagnosed breast cancer cases. Aromatase, a key enzyme in estrogen biosynthesis, represents a well-validated therapeutic target, and indole derivatives have emerged as promising candidates, exhibiting potent activity in the nanomolar concentration range. An integrated computational strategy was employed to gain deeper insights into the molecular basis of aromatase inhibition by indole analogs. The approach combined molecular docking, Self-Organizing Molecular Field Analysis (SOMFA)-based 3D quantitative structure–activity relationship (3D-QSAR) modeling, pharmacophore identification, and 100-nanosecond molecular dynamics (MD) simulations. Docking results highlighted that compound 4, the most potent molecule in the dataset, demonstrated stronger binding interactions within the aromatase active site than the clinically approved reference drug letrozole. The 3D-QSAR SOMFA model effectively captured key molecular features—specifically shape and electrostatic field contributions—that correlate with biological activity. Pharmacophore modeling revealed essential structural characteristics responsible for high inhibitory potency, including one hydrogen bond acceptor and three aromatic moieties. Additionally, MD simulations verified the stability of compound 4’s binding pose within the active site throughout the simulation period, reinforcing its potential as a lead candidate. A newly designed analog, referred to as S8, was computationally predicted to possess a pIC?? value of 0.719 nM, comparable to that of compound 4, suggesting it may serve as an equally effective inhibitor. Overall, these findings offer valuable molecular insights that can facilitate the rational design of next-generation indole-based aromatase inhibitors for the targeted treatment of ER? breast cancer(28).
Fig.6
Jia Cheng Qian et, al., looks at how heat affects the transformation of indole-3-carbinol (I3C), a bioactive molecule produced from glucosinolates found in cruciferous vegetables (CVs) like broccoli. Although I3C is known for its potential health advantages, notably its anti-cancer qualities, little study has been conducted into how cooking impacts its chemical changes. revealed that thermal processing of broccoli florets facilitates the efficient transformation of indole-3-carbinol (I3C) into various biologically active indole derivatives through N-indolylmethylation and N-hydroxymethylation reactions. These chemical conversions are mediated by reactive intermediates such as 3-methyleneindole and formaldehyde, which are generated in situ—3-methyleneindole forms via dehydration of I3C, while formaldehyde arises during the dimerization of I3C into 3,3′-diindolylmethane (DIM). One of the key transformation products, N-(indol-3-ylmethyl)-3,3′-diindolylmethane (DIM-1), was quantified in steamed broccoli florets at concentrations ranging from 0.4 to 4.6?µg per gram. Biological evaluation of DIM-1 demonstrated potent anticancer activity, particularly against A375 human melanoma cells, where it exhibited an IC?? value of 1.87?µM. These findings underscore the role of cooking in enhancing the formation of health-promoting indole derivatives from dietary I3C and highlight DIM-1 as a promising candidate for anticancer drug development. Overall, the study broadens our understanding of I3C’s metabolic transformations and reinforces the therapeutic relevance of its derivatives in cancer prevention and treatment (29).
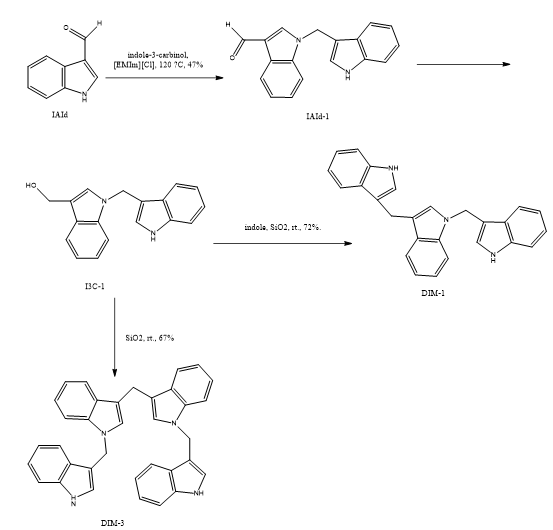
Scheme 22
Jinxuan Li et, al., addressed the urgent need for new antibacterial medicines by developing and synthesizing a novel family of Methicillin-resistant Staphylococcus aureus strain 31 (also called MRSA and other multidrug-resistant microbes are targeted by indole-benzosulfonamide oily acid (OA) analogues 75. To increase antibacterial action, the pharmacophores benzosulfonamide and indole were semisynthetically incorporated into the Op structure in this study. When compared to the traditional antibiotic norfloxacin, metabolite c17 showed the strongest antibacterial activity in vitro against Staphylococcus strains, including MRSA, out of the 26 compounds that were synthesized. In a mouse skin model infected with MRSA, C17 demonstrated remarkable efficacy, significantly reducing the bacterial burden. According to mechanistic studies, c17 may prevent and stop the formation of biofilms while also jeopardizing the functionality of the membranes of bacteria, two crucial strategies for getting past resistance. Additionally, c17 showed a good safety profile by exhibiting little blood coagulation and negligible damage to animal lines of cells (NIH 3T3 and HEK 293T). Given these promising results, indole-benzosulfonamide OA derivatives—in particular, c17—are prime candidates for further study as anti-MRSA therapeutic agents (30).
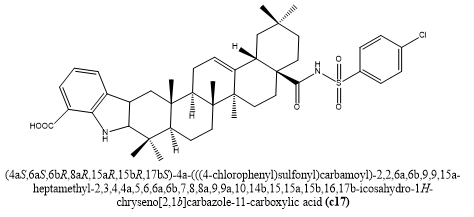
Fig.7
Kamalpreet Kaur et, al., developed a novel class of indole–oxadiazole hybrid molecules was synthesized and investigated for their potential as therapeutic agents against estrogen receptor-positive (ER?) breast cancer. These compounds were evaluated for their cytotoxic effects against two ER-α–positive breast cancer cell lines, T-47D and MCF-7. The results revealed significant antiproliferative activity, with IC?? values ranging between 1.78 and 19.74?μM. Notably, several derivatives—including 5a, 5c, 5e through 5h, and 5j through 5o—exhibited superior inhibitory effects against T-47D cells compared to the standard selective estrogen receptor modulator (SERM), bazedoxifene. Among the tested compounds, 5o emerged as the most potent, demonstrating a strong anti-proliferative effect with an IC?? of 1.72?±?1.67?μM and a remarkably high binding affinity to ER-α (213.4?pM), surpassing bazedoxifene by over 1,500-fold. Compound 5c also showed notable performance, exhibiting an IC?? of 3.24?±?0.46?μM and a binding affinity of 446.6?nM. Western blot analyses confirmed that both 5c and 5o effectively suppressed ER-α protein expression and interfered with its downstream signaling pathways, supporting their role as antagonists. Molecular docking studies revealed that these compounds induced significant conformational alterations in the ER-α receptor, consistent with their antagonistic mechanism of action. Additionally, in silico pharmacokinetic profiling indicated favorable drug-like properties. Molecular dynamics simulations and density functional theory (DFT) calculations further validated their structural stability, chemical reactivity, and potential biological effectiveness. Together, these findings suggest that compounds 5c and 5o are promising leads for the development of new ER-α–targeted therapies for the treatment of hormone-dependent breast cancers(31).
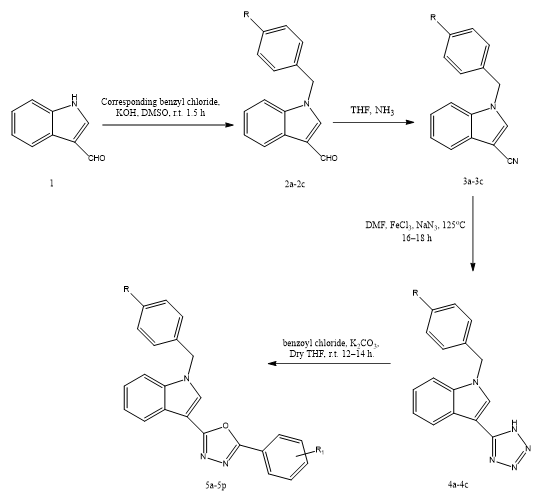
Scheme 23
Sicong Wang et al. concentrated on antioxidant mechanisms to aid in cellular defense against elevated oxygen species that are reactive (ROS), such as the pathway of glutathione (GSH) and a protein known as (Trx) pathways, which are commonly overexpressed in cancers like lymphoma. Although metal-based substances like auranofin have demonstrated potential in focusing on enzymes that contain thiols and selenium, such as thioredoxin reductase (TrxR), their clinical application has been constrained by issues like resistance, toxicity, and poor selectivity. Eleven new indole-metal complexes were developed for this study, and their anti-inflammatory properties were evaluated in cancer cell lines. Associations based on indole-gold(I) functioned better against lymphoma than those based on iron and cobalt because they inhibited the activity of glutathione peroxidase (Gpx) and TrxR, but not the enzyme glutathione reductase (GR).Notably, it was found that complexes 3h and 3i cause ferroptosis, as evidenced by lipid oxidation and elevated expression of enzymes linked to iron metabolism. Significant anti-angiogenic effects were shown in living cells using zebrafish embryos, which prevented the formation of new blood vessels. Overall, our research shows that 3h and 3i are potent dual TrxR and Gpx antagonists that can cause ferroptosis, which makes them attractive options for the creation of anticancer drugs that target lymphoma (32).
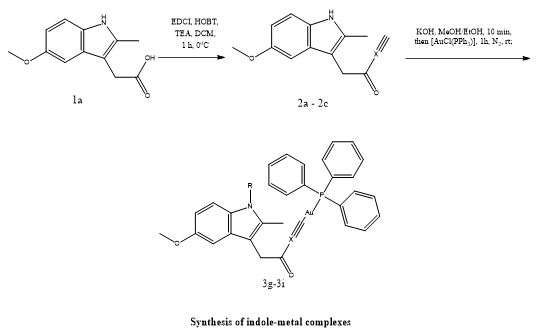
Scheme 24
Yareeb J. Sahar's et, al., focused on the development and characterization of a novel azo dye 12 ligand, (HIDAT), produced by reacting indole with the 12-diazonium ion of 2-aminothiazole. Utilizing a variety of methods, such as elemental analysis (C.H.N.S.), mass spectrometry, ~1H NMR, FT-IR, UV-visible spectroscopy, magnetic susceptibility, and 12molar conductivity, the HIDAT ligand and its transition metal 12complexes with cobalt, copper, and nickel and palladium were successfully synthesized and defined. The palladium (II) complex was created in a 1:1 ratio, but the other complexes were synthesized in a 1:2 metal-to-ligand ratio. Spectroscopic data indicated that HIDAT operates as a bidentate ligand, implying octahedral geometry for all metal complexes except the palladium complex, which has a square planar structure. HIDAT also demonstrated pH sensitivity in aqueous solutions. Cytotoxicity experiments revealed that the palladium complex was much more harmful to human leukemia HL-60 cells than to normal HdFn cells. Furthermore, molecular docking (MOE) investigations revealed that the palladium complex had a considerable inhibitory effect on the leukemia cancer target protein (3s7s), confirming its potential as an anticancer treatment(33).
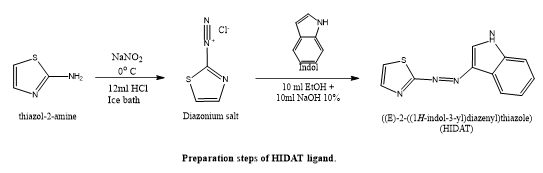
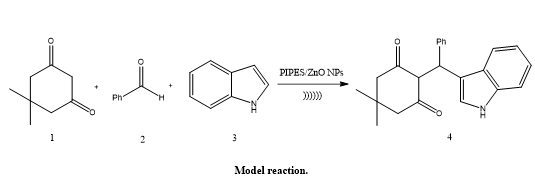
Suchita S. Gadekar described the Zinc oxide nanoparticles (ZnO NPs) and 1,4-piperazine diethanesulfonic acid (PIPES) were synthesized, characterized, and used catalytically as a dual catalytic system by Suchita S. Gadekar. Under ultrasonic irradiation at 60°C, this catalyst effectively promoted the reaction of dimedone, benzaldehyde, and indole to generate 2-((1H-indol-3-yl)(phenyl)methyl)-5,5-dimethylcyclohexane-1,3-dione. The catalytic performance was greatly improved by the combination of PIPES's Brønsted acidity and ZnO NPs' surface-active characteristics. The produced molecule also has medicinal relevance, which emphasizes the benefits of this green synthesis method with ultrasonic assistance.(34).
Scheme 25
Concluding Remarks
n conclusion, this study offers a thorough description of the most recent developments (2020–2025) in the synthesis, pharmacological evaluation, and modification of indole and its analogs. Because of its wide range of biological activities, ease of structural modification, and chemical versatility, indole continues to be a highly significant heterocyclic framework in medicinal chemistry. It plays a crucial role in the creation of new therapeutic agents by serving as a fundamental component of biomolecules like melatonin or serotonin as well as being an ingredient in a number of licensed medications, including indomethacin, delavirdine, and Panobinostat.(35). The studies reviewed herein span a diverse array of biological applications, including anticancer, antimicrobial, anti-inflammatory, antioxidant, antidiabetic, and anti-leishmanial activities. Importantly, several indole derivatives exhibited significant potency against multidrug-resistant pathogens and cancer cell lines, highlighting their potential to overcome major obstacles in current therapeutic regimens. Structure–activity relationship (SAR) analyses, molecular docking studies, and mechanistic insights have deepened our understanding of the interaction between indole frameworks and their biological targets, aiding in rational drug design and lead optimization. The synthetic approaches described from classical methods like Fischer and Madelung indole syntheses to green, one-pot, and microwave-assisted strategies underscore the ongoing innovation in constructing structurally diverse indole derivatives with improved pharmacological profiles. These environmentally friendly and efficient methods align with the principles of sustainable chemistry and provide powerful tools for future medicinal chemistry endeavors. Despite the substantial progress made, it is evident that the full potential of indole chemistry remains underexploited. Many promising leads await further in vivo validation, toxicity profiling, and clinical translation. Moreover, the design of multifunctional or multitarget-directed ligands (MTDLs) based on the indole scaffold offers an exciting avenue for addressing complex and multifactorial diseases such as cancer, neurodegeneration, and emerging viral infections. This review aims to bridge the existing gaps in the literature by providing an integrated and up-to-date account of indole-based drug discovery. It serves not only as a valuable resource for chemists, pharmacologists, and drug developers but also as an inspiration for future interdisciplinary research. With continued advancements in synthetic methodologies, computational tools, and biological screening platforms, the indole scaffold is poised to drive the next wave of innovation in therapeutic development. The pressing global need for novel and effective agents makes this an opportune time to further explore and harness the vast therapeutic potential of indole-based compounds.
REFERENCES


Manish Kathiriya*, Bhavesh Akbari, Pradeep Kumar M. R., Indole: A Potent Scaffold with Versatile Pharmacological Activities, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 309-344. https://doi.org/10.5281/zenodo.16737603
 10.5281/zenodo.16737603
10.5281/zenodo.16737603