
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Guru Nanak College of Pharmaceutical Sciences, Dehradun
This comparative study examines drug recall procedures in India and the United States, focusing on regulatory evolution, classification systems, procedural frameworks, and enforcement mechanisms. Both nations developed their drug regulatory systems in response to public health catastrophes—India’s Maselkar poisonings (1937) leading to the Drugs and Cosmetics Act (1940) and Rules (1945), and America’s Elixir Sulfanilamide disaster (1937) prompting the Federal Food, Drug, and Cosmetic Act (1938), later strengthened by the Kefauver Harris Amendments (1962). While India’s Central Drugs Standard Control Organization (CDSCO) operates within a federal structure shared with state authorities, the U.S. Food and Drug Administration (FDA) functions as a centralized, unified regulator. Both systems classify recalls into three tiers (Class I – most serious, Class II – moderate, Class III – minor), but differ in terminology, risk assessment methods, and transparency mechanisms. India mandates faster notification timelines (24 hours for Class I) and requires annual mock recalls, whereas the FDA employs a more structured Health Hazard Evaluation (HHE) and publishes weekly Enforcement Reports. The CDSCO possesses broader unilateral recall powers (Section 26A), while the FDA must seek court orders for mandatory recalls. Key recommendations for India include enacting a standalone recall law, harmonizing state central coordination, investing in digital traceability, and integrating pharmacovigilance. For the U.S., expanding unannounced international inspections, enhancing real time supply chain monitoring, and leveraging Quality Management Maturity (QMM) programs are advised. Global convergence through ICH guidelines and WHO alert platforms is essential for mutual recognition and patient safety.
Historical Evolution and Regulatory Framework
The evolution of drug regulation in India represents a fascinating journey from colonial legacy to contemporary modernization, beginning with the Drugs and Cosmetics Act of 1940, which was subsequently followed by the Drugs and Cosmetics Rules of 1945. This foundational legislation emerged in response to a tragic incident known as the Maselkar poisonings of 1937, where a contaminated liquid caused numerous fatalities, compelling the British colonial administration to establish formal controls over drug quality and safety[1]. The Act originally focused primarily on prohibiting the import, manufacture, distribution, and sale of substandard and misbranded drugs, with its scope gradually expanding through numerous amendments to encompass cosmetics, Ayurvedic, Siddha, and Unani medicines. The accompanying Rules of 1945 provided the operational machinery, detailing specific requirements for licensing, inspection, sampling, and analytical testing, thereby creating a comprehensive administrative framework that has served as the backbone of Indian pharmaceutical regulation for over eight decades. The Central Drugs Standard Control Organization (CDSCO) emerged as the apex regulatory authority under these provisions, with its role evolving significantly from a relatively peripheral coordinating body to the central pillar of drug oversight in India. Headquartered in New Delhi with zonal offices across the country, the CDSCO is responsible for approving new drugs, conducting clinical trial oversight, establishing quality standards for drugs and cosmetics, coordinating the work of state drug control authorities, and regulating imports of pharmaceutical products[2]. The organization operates under the Directorate General of Health Services within the Ministry of Health and Family Welfare, and its responsibilities have expanded considerably to include the regulation of medical devices, biological products, and vaccines, particularly following India's emergence as a major pharmaceutical manufacturing hub serving both domestic and international markets. The most recent significant development in Indian drug regulation came with the Schedule M amendments of 2023, which represented a transformative overhaul of Good Manufacturing Practices (GMP) requirements for pharmaceutical manufacturers[3]. These amendments radically upgraded manufacturing standards to align more closely with World Health Organization guidelines and international benchmarks such as those of the European Union and the United States Food and Drug Administration, introducing stringent requirements for premises design, equipment qualification, quality management systems, self-inspection protocols, validation of critical processes, and documentation practices. The revised Schedule M mandates the implementation of quality risk management principles, requires comprehensive environmental monitoring for sterile product manufacturing, imposes stricter controls on cross-contamination prevention, and establishes enhanced requirements for computer system validation in automated manufacturing environments[4]. Small and medium-sized enterprises were granted extended timelines to achieve compliance, but the overall direction is unmistakably toward harmonization with global regulatory standards, reflecting India's ambition to strengthen its position as a leading supplier of high-quality generic medicines to markets worldwide. In parallel, the evolution of drug regulation in the United States followed a distinct but equally significant trajectory, anchored by the Federal Food, Drug, and Cosmetic (FD&C) Act of 1938, which replaced the earlier Pure Food and Drug Act of 1906 after a public health tragedy of enormous proportions. The Elixir Sulfanilamide disaster of 1937, in which a diethylene glycol-based liquid formulation caused over one hundred deaths, many of them children, galvanized public opinion and legislative action, leading to the enactment of the FD&C Act that fundamentally transformed American drug regulation. Unlike its predecessor, which primarily addressed adulteration and misbranding after products reached the market, the 1938 Act required premarket notification for new drugs, mandated that manufacturers provide evidence of safety before marketing, authorized factory inspections, established standards for food and cosmetics, and provided the government with court-ordered injunctions and seizures as enforcement tools[5][6]. The Act has been strengthened through numerous amendments over the ensuing decades, most notably the Kefauver-Harris Amendments of 1962, which were precipitated by the thalidomide tragedy that caused devastating birth defects in thousands of children born to mothers who had taken the drug during pregnancy.
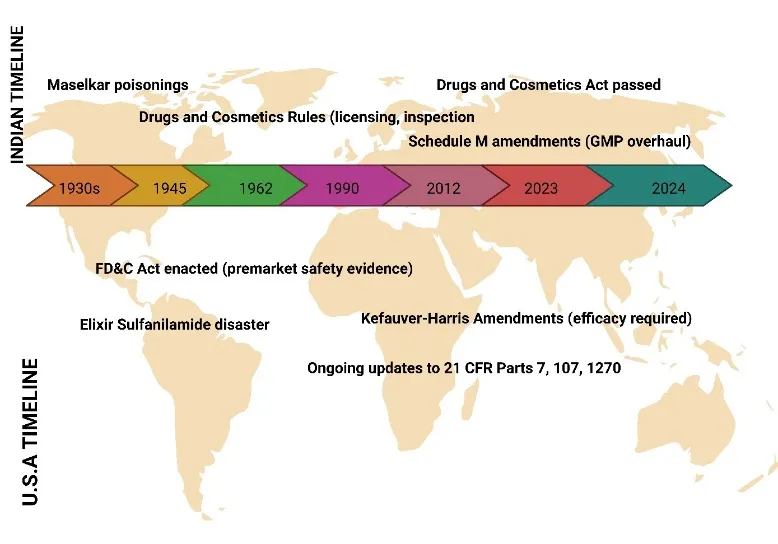
Fig: 1 Timeline of Key Regulatory Events in Drug Safety for India and the USA
These amendments introduced the revolutionary requirement that drug manufacturers must prove not merely safety but also efficacy through substantial evidence derived from adequate and well-controlled clinical investigations, fundamentally shifting the regulatory paradigm from a purely safety-focused system to one demanding demonstrated therapeutic benefit[7][8]. The United States Food and Drug Administration (FDA) serves as the primary enforcement agency for the FD&C Act and has evolved from a small bureau of chemists in the early twentieth century to a sophisticated scientific and regulatory agency employing over 18,000 personnel, including physicians, pharmacologists, chemists, microbiologists, statisticians, and inspectors. The FDA's Center for Drug Evaluation and Research (CDER) oversees the approval of new pharmaceutical products, conducting rigorous reviews of clinical trial data, manufacturing processes, labeling, and proposed post-marketing surveillance plans, while the Office of Regulatory Affairs manages field operations including inspections of domestic and foreign manufacturing facilities. The FDA operates under a risk-based regulatory philosophy, allocating resources to areas of highest public health concern, and has developed increasingly sophisticated tools including expedited approval pathways for breakthrough therapies, adaptive trial designs, real-world evidence frameworks, and patient-focused drug development initiatives that incorporate patient experience data into regulatory decision-making[9][10]. Key regulations under Title 21 of the Code of Federal Regulations (CFR) provide the detailed operational framework for FDA enforcement, with Parts 7, 107, and 1270 addressing particularly critical aspects of the agency's authority. Part 7 establishes the comprehensive regulatory framework for enforcement actions, including the procedures for seizure, injunction, and prosecution of violative products, the criteria for determining when products are considered adulterated or misbranded, and the guidelines for voluntary recall procedures that enable manufacturers to remove problematic products from commerce without formal legal proceedings[11][12]. Part 107 specifically addresses biological products, including the licensing requirements for manufacturers, establishment standards for facilities producing biological drugs, blood and blood component regulations, and the donor eligibility and testing protocols designed to minimize the risk of infectious disease transmission through biological therapeutics. Part 1270 governs human tissue intended for transplantation, establishing donor screening and testing requirements, recordkeeping protocols that enable traceability from donor to recipient, and quarantine and disposition procedures for tissues that fail to meet regulatory standards, thereby addressing the unique risks associated with cellular and tissue-based products that cannot be sterilized through conventional terminal processing methods[13].
A comparative summary of these regulatory milestones reveals both striking convergences and instructive divergences between the Indian and American systems, with each nation's approach reflecting its unique historical, political, and industrial circumstances. Both regulatory frameworks originated in response to specific public health catastrophes that captured public attention and created political momentum for legislative action, with India's Maselkar poisonings and America's Elixir Sulfanilamide disaster serving as similar catalysts that transformed reactive, post-market regimes into more proactive, pre-market oversight systems[14][15]. However, the timing and pace of evolution differ dramatically, with the United States establishing its foundational modern framework in 1938 and adding the efficacy requirement in 1962, while India's regulatory system remained relatively static for much of the twentieth century before accelerating its modernization efforts in the early 2000s in response to export market requirements and World Trade Organization commitments. The institutional structures also reveal interesting contrasts, as the FDA operates as a unified, centralized federal authority with direct enforcement jurisdiction across all fifty states, while India's CDSCO functions within a more complex federal structure where state drug control authorities retain primary responsibility for licensing and inspecting manufacturing facilities within their territories, with the central authority playing predominantly coordinating and standard-setting roles[16]. The recent Schedule M amendments in India represent a significant effort to reduce this regulatory fragmentation and strengthen quality assurance, effectively mirroring the GMP requirements that have been enforced in the United States for decades under 21 CFR Parts 210 and 211. Despite these differences, both systems increasingly recognize the imperatives of international regulatory convergence, particularly as global supply chains become more integrated and pharmaceutical products routinely cross multiple national borders before reaching patients[17][18]. India has participated actively in the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and has sought mutual recognition agreements with major regulatory bodies, including the FDA, while the United States has increased its inspection presence in India and other developing countries through expanded foreign inspection programs and information-sharing arrangements. The fundamental objectives of both regulatory systems remain remarkably aligned despite their different historical trajectories: ensuring that pharmaceutical products are safe, effective, and of consistent quality, maintaining the integrity of clinical research data, protecting patients from counterfeit and substandard medicines, and fostering an environment that encourages innovation while maintaining rigorous public health protections. As pharmaceutical science continues to advance with breakthroughs in biologics, gene therapies, personalized medicines, and digital health technologies, both India and the United States face the ongoing challenge of adapting their regulatory frameworks to address novel risks while avoiding unnecessary barriers to patient access, ensuring that the evolution of drug regulation remains responsive to both scientific progress and public health imperatives[19].
Drug Recall Classification Systems
The Indian Classification System
Class I
In the Indian regulatory framework, which is primarily governed by the Drugs and Cosmetics Act, 1940 and Rules, 1945 along with the recent guidance on recall procedures issued by the Central Drugs Standard Control Organization (CDSCO), drug recalls are classified into three categories based on the potential severity of the associated health hazard. Class I, designated as “Critical,” represents the highest level of urgency and applies to situations where there is a reasonable probability that the use of or exposure to a defective drug product will cause serious adverse health consequences or death[20]. This classification is reserved for products with critical defects such as contamination with highly toxic substances (e.g., bacterial endotoxins, heavy metals, or cross-contamination with potent cytotoxic drugs), incorrect active pharmaceutical ingredient (API) content leading to overdose or underdose of a narrow therapeutic index drug, complete lack of sterility assurance in a parenteral product, or packaging mix-ups that result in life-threatening medication errors. Under Indian guidelines, a Class I recall is triggered when a manufacturer or the CDSCO identifies a defect that is life-threatening or likely to cause a serious, permanent impairment of body function. The process mandates immediate public notification through press releases, direct communication with healthcare professionals and hospitals, and a request for the return of all distributed batches down to the patient level. The CDSCO expects manufacturers to initiate a Class I recall within 24 hours of defect confirmation, with daily status reports submitted to the regulatory authority until the recall is completed[21].
Class II
Class II in the Indian system is termed “Major” and encompasses situations where the product defect may cause temporary or medically reversible adverse health consequences, or where the probability of serious adverse events is remote but not negligible. This classification typically includes defects such as minor deviations from approved specifications that do not compromise safety in a critical manner, for example, a slight variation in dissolution rate that remains within pharmacopoeial limits for most but not all batches, or labeling errors that omit a non-serious warning or provide incorrect but non-dangerous dosage instructions. Other examples include the presence of a foreign particulate matter that is not pyrogenic or toxic, a modest potency variation that does not push the drug outside its therapeutic window, or the absence of a stability-indicating parameter that might affect shelf-life but not immediate safety[22][23]. Under Indian recall procedures, a Class II recall requires the manufacturer to notify the CDSCO and all direct consignees (wholesalers, hospitals, and major retailers) within 72 hours of defect determination, but public notification through mass media is generally not required unless the defect becomes widespread. The manufacturer is expected to retrieve the affected product from the distribution chain, though retrieval down to the individual patient level is not mandated unless the defect is discovered after dispensing. The CDSCO monitors Class II recalls through periodic progress reports, and the recall is considered complete when the manufacturer demonstrates that 95% or more of the distributed product has been returned or accounted for, with a detailed root-cause analysis and corrective action preventive action (CAPA) plan submitted for approval[24].
Class III
Class III, designated as “Minor,” applies to product defects that are unlikely to cause any adverse health consequence, even in sensitive or vulnerable patient populations. This classification is used for violations of labeling regulations that do not affect patient safety, such as a missing “caution: federal law prohibits dispensing without prescription” statement that is not legally required in India but may appear on export products, or an incorrect batch number printed on a secondary carton that does not affect traceability. Other examples include the presence of a harmless cosmetic defect like a slightly discolored tablet coating with no impact on stability or efficacy, a missing tamper-evident seal on an over-the-counter product where the seal is not required by regulation, or the absence of a non-critical excipient declaration on the package insert[25][26]. Under Indian guidelines, a Class III recall does not require immediate action; manufacturers are typically given 30 days to notify the CDSCO and voluntarily remove the affected product from the market at their own pace. There is no mandatory requirement for direct customer notification, and most Class III recalls are handled through routine inventory reconciliation during the next stock rotation. The manufacturer must still file a recall report with the CDSCO detailing the nature of the defect, the quantity distributed, and the number of units recovered, but no public disclosure is required unless the defect persists across multiple batches or indicates a systemic quality system failure. The CDSCO uses Class III recall data primarily for regulatory intelligence and for prioritizing future inspections, rather than for immediate enforcement action[27].
Timelines for Each Class
The CDSCO has established specific timelines for recall initiation and completion that vary directly with the severity classification. For a Class I (Critical) recall, the manufacturer must notify the CDSCO and all direct consignees within 24 hours of confirming the defect, and a public warning (press release, website notification, or direct healthcare professional communication) must be issued within 48 hours. The recall effectiveness checks must be conducted weekly, and the entire recall process—from initial notification to retrieval of at least 90% of distributed product—should ideally be completed within two weeks, though complex recalls involving international distribution may be extended to 30 days with prior CDSCO approval. For a Class II (Major) recall, notification to the CDSCO and direct consignees must occur within 72 hours of defect confirmation, and the recall is expected to reach 95% completion within 30 to 45 days, depending on the product’s distribution network. Weekly progress reports are required for the first month, followed by monthly reports until closure[29][30]. For a Class III (Minor) recall, the manufacturer has up to 30 days to notify the CDSCO, and the recall can be conducted in the normal course of business without a fixed completion deadline, though the CDSCO expects a final report within six months. In all cases, the manufacturer must submit a final recall report to the CDSCO’s zonal office, including a root-cause analysis, a description of corrective actions taken, and documentation of product disposition (destruction or reconditioning). Failure to adhere to these timelines can result in penalties, suspension of manufacturing licenses, or public naming of the non-compliant manufacturer[31].
The U.S. Classification System
Class I
Under the U.S. Food and Drug Administration (FDA) framework, as defined in 21 CFR Part 7 (Enforcement Policy), recalls are classified into three similarly named but distinctly defined categories based on the level of health hazard presented by the violative product. Class I is the most serious classification and applies when there is a reasonable probability that the use of or exposure to a violative product will cause serious adverse health consequences or death. Unlike the Indian system, the FDA explicitly ties Class I to the concept of “reasonable probability” derived from the Federal Food, Drug, and Cosmetic Act, requiring a scientific assessment of the likelihood of harm, not merely the possibility. Examples of Class I recalls in the U.S. include a blood pressure medication contaminated with a potential carcinogen such as N-nitrosodimethylamine (NDMA) above the acceptable intake limit, intravenous fluids found to be non-sterile leading to risk of bloodstream infections, or a critical dosing error caused by a mislabeled vial where a concentrated solution is packaged as a dilute solution[32]. The FDA mandates that for a Class I recall, the recalling firm must notify the FDA’s district office within 24 hours of determining that a recall is necessary, and the FDA will issue a public press release, update its weekly Enforcement Report, and often post a recall notice on its website and social media channels. The FDA also expects the firm to reach consumers directly when the product has been dispensed to individual patients, for example, through letters to pharmacies and healthcare providers, or even direct patient notification if traceability allows. Class I recalls are treated with the highest enforcement priority, and the FDA may conduct for-cause inspections and seize product if the firm fails to cooperate.
Class II
Class II in the U.S. system is defined as a situation where the use of or exposure to a violative product may cause temporary or medically reversible adverse health consequences, or where the probability of serious adverse health consequences is remote. This classification bridges the gap between life-threatening defects and minor violations. Typical Class II examples include a drug product that fails dissolution specifications but still releases the API sufficiently to achieve therapeutic blood levels in most patients, a minor labeling error such as omitting a non-critical precaution (e.g., “may cause drowsiness” for an antihistamine), or a small potency deviation (e.g., 95% instead of 100% label claim) where the therapeutic window is wide. The FDA requires that for a Class II recall, the firm notify the FDA within 48 to 72 hours, and the FDA will typically issue a press release but only if the product is widely distributed or the defect is likely to cause patient confusion. Unlike Class I, direct patient notification is rarely required for Class II recalls; instead, notification is limited to wholesalers, distributors, and healthcare facilities. The recall effectiveness checks are conducted by the FDA at a lower frequency, and the firm is expected to complete the recall within 30 to 60 days depending on the complexity of the supply chain. The FDA’s Enforcement Report will list the recall as Class II, and the classification can be upgraded or downgraded based on new information received during the recall process[33].
Class III
Class III applies to violative products that are not likely to cause adverse health consequences. This category includes technical violations of FDA regulations that do not pose a direct safety risk, such as a drug product where the packaging does not meet child-resistant closure standards but the product itself is not toxic, or a labeling error that involves a non-meaningful typographical mistake (e.g., misspelling of an excipient name). Other examples include a cream that fails to meet its viscosity specification but remains stable and effective, or a tablet that has a minor surface defect without any impact on dissolution or bioavailability. For a Class III recall, the FDA does not require a press release or public notification unless the violating product is also subject to other enforcement actions. The firm must still notify the FDA district office and submit a recall strategy, but the timeline is flexible, typically allowing 30 to 90 days for completion. The FDA may also allow the firm to conduct a “market withdrawal” instead of a formal recall if the product is not in violation of the FD&C Act but is removed for other reasons (see below). Class III recalls are often resolved through routine returns and credits without any direct patient or provider communication, and they rarely attract media attention or regulatory fines unless they indicate a pattern of poor quality.
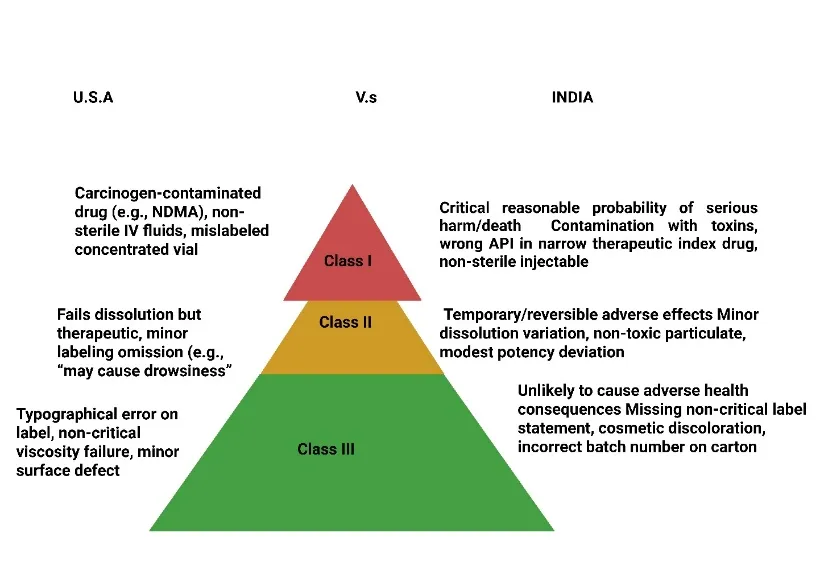
Fig: 2 Risk Classification of Pharmaceutical Products in the U.S. and India
Market Withdrawal vs. Recall
A critical distinction in the U.S. system that has no exact parallel in the Indian framework is the difference between a “market withdrawal” and a formal “recall[34][35].” A market withdrawal occurs when a firm removes a product from the market for reasons that do not violate the FD&C Act, such as a cosmetic defect (e.g., off-color appearance with no safety impact), a change in product formulation that makes the old version obsolete, or a minor packaging change that does not affect safety or efficacy. The key legal distinction is that a market withdrawal is not a recall because there is no violation of law, and therefore the FDA does not classify it, does not list it in the Enforcement Report, and does not require the firm to submit a recall strategy or effectiveness checks. In contrast, a recall is always based on a violation of the FD&C Act or related regulations, and the FDA assigns a classification (I, II, or III) based on health hazard evaluation. This distinction is important for manufacturers because recalls carry regulatory consequences, including increased inspection scrutiny, potential warning letters, and reputational damage, whereas market withdrawals are considered routine business decisions. However, the FDA reserves the right to reclassify a market withdrawal as a recall if it determines that a violation actually exists. For example, if a firm withdraws a drug due to “off-odor” but the odor is later traced to a degradation product that could be toxic, the FDA may retroactively classify it as a Class I or II recall. Pharmaceutical companies are therefore required to report any product removal to the FDA within 24 hours if there is any possibility of a violation, even if they initially believe it is a market withdrawal[36].
Comparative Analysis: Classification Criteria and Severity Assessment
A systematic comparison of the Indian and U.S. drug recall classification systems reveals significant convergences in the three-tiered structure but important divergences in the operational definitions, assessment methodologies, and enforcement mechanisms. Both systems recognize three severity levels – Class I (most severe), Class II (moderate), and Class III (minor) – and both systems base classification on the probability and seriousness of adverse health consequences. However, the Indian system’s labels (“Critical,” “Major,” “Minor”) are more descriptive of the defect itself, while the U.S. system’s labels (“Most Serious,” “Moderate Risk,” “Lowest Risk”) focus on the patient outcome risk. In terms of triggering criteria, the U.S. FDA employs a more explicit risk assessment framework, often using quantitative methods such as fault tree analysis, hazard analysis critical control points (HACCP), and probabilistic risk assessment to determine whether the probability of harm is “reasonable[37][38].” India’s CDSCO, while moving toward risk-based regulation, still relies largely on expert judgment and precedent-based classification, which can lead to variability between different zonal offices. For example, the same defect – a 10% over-potency in a drug with a wide therapeutic window – might be classified as Class II by one Indian state authority but as Class III by another, whereas the FDA has published guidance documents that provide more specific examples and algorithms. Another major difference lies in the timeline expectations: India mandates faster action for Class I recalls (24-hour notification) compared to the U.S. (also 24 hours, but with more stringent requirements for public disclosure). However, the U.S. system has a more robust mechanism for recall effectiveness checks, requiring firms to verify that each consignee actually received and acted upon the recall notice, with a target effectiveness level of 100% for Class I recalls[39][40]. India’s effectiveness target for Class I is 90%, reflecting the practical challenges of tracking products in a fragmented supply chain with many small retailers. The U.S. distinction between market withdrawal and recall is a notable feature absent from Indian regulations, where any product removal due to a defect is typically processed as a recall, regardless of whether a legal violation exists. This can lead to over-reporting in India but also reduces the risk of firms incorrectly labeling a dangerous defect as a non-violative withdrawal. In terms of severity assessment, both systems consider similar factors: the nature and severity of the hazard (e.g., toxicity, contamination, mislabeling), the duration of exposure, the number of patients affected, and the vulnerability of the patient population (pediatrics, geriatrics, immunocompromised). However, the U.S. FDA places greater emphasis on causal evidence and the “reasonable probability” standard, which requires a showing that harm is likely to occur, not merely possible. India’s standard is less explicitly defined, often relying on the manufacturer’s self-assessment or the CDSCO’s prima facie judgment. Another convergence is the increasing use of electronic recall management systems: the FDA uses the Recall Enterprise System (RES) for real-time tracking, while CDSCO is piloting a similar digital portal for recall notifications. Both regulators also share information through international mechanisms such as the International Coalition of Medicines Regulatory Authorities (ICMRA) and the World Health Organization’s Global Surveillance System for substandard and falsified medical products. Ultimately, while the Indian system is rapidly harmonizing with global best practices – especially after the 2023 Schedule M amendments that emphasize risk-based quality management – the U.S. system remains more mature in terms of standardized classification criteria, transparency through public enforcement reports, and legal remedies for consumers affected by defective products. Nevertheless, both systems face common challenges: ensuring timely classification in the face of incomplete data, preventing under-classification due to industry pressure, and maintaining public trust when recalls are announced. As pharmaceutical supply chains become increasingly global, the convergence of recall classification criteria between India and the U.S. will be essential for mutual recognition of recall actions and for protecting patients who may receive the same defective product across both jurisdictions.
Drug Recall Procedures
The Indian Recall Procedure
Voluntary Initiation by Licensee
Under the Drugs and Cosmetics Act, 1940 and Rules, 1945, the primary responsibility for initiating a recall rests squarely with the licensee, i.e., the manufacturer, importer, or marketing authorization holder. The Indian regulatory framework encourages voluntary recall as the preferred first step, recognizing that manufacturers possess the most detailed knowledge of their products, supply chains, and quality systems[41]. When a licensee discovers or suspects a defect – whether through internal quality testing, post-market surveillance, customer complaints, or adverse event reports – they are expected to conduct an immediate investigation and, if the defect is confirmed, voluntarily initiate a recall without waiting for a directive from the Central Drugs Standard Control Organization (CDSCO). The manufacturer must self-assign a provisional recall classification (Class I, II, or III) based on the severity of the health hazard and then notify the CDSCO’s zonal office and the respective state drug control authority within the timelines prescribed: 24 hours for Class I, 72 hours for Class II, and 30 days for Class III. The notification must include a detailed recall report specifying the product name, batch number, quantity distributed, distribution network details (wholesalers, retailers, hospitals), the nature of the defect, the root cause (if known), and a proposed recall strategy. The manufacturer is also required to designate a recall coordinator who will serve as the single point of contact for regulatory authorities throughout the recall process. Voluntary initiation does not exempt the licensee from subsequent regulatory oversight, but it is viewed favourably by the CDSCO, often resulting in reduced penalties and cooperative compliance treatment. Conversely, failure to voluntarily recall a dangerous product can lead to suspension or cancellation of the manufacturing license, substantial fines, and even criminal prosecution under Section 27 of the Drugs and Cosmetics Act[42].
Statutory Powers of CDSCO
While voluntary recall is the cornerstone of the Indian system, the CDSCO possesses significant statutory powers to compel recalls when a licensee is unwilling, unable, or unreasonably slow to act. These powers derive primarily from the Drugs and Cosmetics Act, particularly Sections 26A (power to prohibit manufacture and sale of drugs in public interest) and 22 (powers of inspectors to seize samples and suspend licenses)[48]. The CDSCO can issue a formal “recall order” directing the manufacturer to immediately stop distribution, notify all consignees, and retrieve the affected product. This order can be issued based on its own inspection findings, complaints received from healthcare professionals or patients, or intelligence from international regulators. If the manufacturer fails to comply, the CDSCO can authorize its zonal or sub-zonal officers to enter the manufacturer’s premises, seize all remaining stock, and even destroy the product at the manufacturer’s expense[49]. Additionally, the CDSCO can publicly disclose the recall and the manufacturer’s non-cooperation through its website and press releases, which carries severe reputational risk. In extreme cases where a defective product poses an imminent threat to public health (e.g., a contaminated intravenous fluid already distributed to multiple hospitals), the CDSCO can invoke emergency provisions to coordinate with state drug controllers for a state-wide or nationwide product seizure without prior notice to the manufacturer. The statutory powers also extend to importers: the CDSCO can suspend an import license and direct customs authorities to block further shipments of a recalled product at all ports of entry. Despite these broad powers, the CDSCO historically used them sparingly, preferring voluntary compliance. However, following the 2023 Schedule M amendments and the revised recall guidelines of 2024, the CDSCO has adopted a more assertive stance, issuing several recall orders against manufacturers found to have shipped grossly substandard drugs, especially those destined for export markets[50].
The "Recall and Rapid Alert System" (2012, revised 2024)
The operational backbone of India’s recall framework is the “Recall and Rapid Alert System,” originally introduced in 2012 and substantially revised in 2024 to address gaps in speed, transparency, and international alignment. This system establishes a centralized digital portal (the National Drug Recall Portal) where all recall notifications, progress reports, and final closure documents must be filed by manufacturers. The 2024 revision introduced several critical enhancements: first, it mandates a 24/7 rapid alert communication channel for Class I recalls, requiring manufacturers to send text alerts and emails to all consignees within six hours of CDSCO confirmation. Second, it created a three-tier rapid alert escalation protocol – Level 1 for state-level recalls, Level 2 for regional recalls (multiple states), and Level 3 for nationwide recalls – with each level triggering progressively broader public notification requirements[51]. Third, the revised system integrates with the World Health Organization’s Global Surveillance System, requiring the CDSCO to report any Class I recall involving an exported product to the importing country’s regulator within 48 hours. Fourth, the system now requires manufacturers to upload electronic copies of batch manufacturing records, distribution logs, and quality test results to the portal within 24 hours of initiating a recall, enabling the CDSCO to conduct real-time verification. The “Rapid Alert” component is specifically designed for situations where a defect is discovered after the product has reached consumers; it requires the manufacturer to immediately issue a “Dear Doctor” letter, a public health advisory, and if necessary, a television or radio warning in regional languages. The 2024 revision also introduced a color-coded dashboard that publicly displays the status of all ongoing recalls – green (compliant, on track), yellow (delayed, under monitoring), red (non-compliant, enforcement action pending) – to increase transparency and peer pressure on manufacturers[52].
Levels of Recall: Consumer, Retail, Wholesale
The Indian system defines three distinct levels of recall based on how far down the distribution chain the manufacturer must retrieve the product. The wholesale level is the shallowest, requiring the manufacturer to notify and retrieve the product only from its direct wholesalers and distributors. This level is typically used for Class III (Minor) defects where the product has not yet reached retail pharmacies or hospitals, or where the defect is purely cosmetic and poses no safety risk. The manufacturer must issue a recall notice to each wholesaler, obtain a signed acknowledgment, and arrange for return or destruction of the recalled stock, with no requirement to contact retail outlets or consumers[53][54]. The retail level goes one step deeper: the manufacturer must notify and retrieve the product from all retail pharmacies, hospital pharmacies, and clinic stores. This level is standard for Class II (Major) recalls where the defect may cause temporary but reversible adverse effects. The manufacturer is expected to provide retailers with prepaid shipping labels or arrange for field representatives to physically collect the recalled product. Additionally, the manufacturer must post a recall notice on its website and, if the product is commonly sold over the counter, issue a public notice in local newspapers. The consumer level is the deepest and most resource-intensive, reserved for Class I (Critical) recalls where the product has already been dispensed to individual patients. At this level, the manufacturer must make “reasonable efforts” to contact each patient who received the product, using pharmacy dispensing records, patient registries, or even media advertisements requesting patients to return the product[55]. The consumer level recall also requires the manufacturer to set up a toll-free helpline, a dedicated recall website, and to coordinate with healthcare providers to arrange for alternative therapy or medical monitoring of affected patients. For example, a recall of a contaminated blood pressure medication would require the manufacturer to work with cardiologists and pharmacies to identify patients who took the affected batch and schedule follow-up blood tests. The CDSCO mandates that for consumer level recalls, the manufacturer must achieve a minimum retrieval rate of 90% of distributed units, or provide a scientifically justified explanation why a lower rate is acceptable (e.g., short-duration use products like antibiotics may have already been consumed)[56].
Mock Recall Requirements
Since the 2023 Schedule M amendments, Indian manufacturers are now required to conduct and document mock recalls (also called simulated recalls) at least once annually for each product category. A mock recall is a planned exercise designed to test the effectiveness of the manufacturer’s recall system, including the accuracy of distribution records, the speed of notification, and the responsiveness of consignees[57]. The manufacturer must simulate a recall of a specific batch from a specific product, tracing the batch from production through distribution to the wholesale or retail level using actual records. The exercise must measure key performance metrics: time taken to identify all consignees, time taken to issue recall notifications (actual letters or emails sent, not hypothetical), percentage of consignees who respond within the target timeframe, and the completeness of the distribution records. The results must be documented in a mock recall report, which is reviewed annually by the CDSCO during inspections. If a manufacturer’s mock recall reveals significant deficiencies – such as missing consignee contact information, inability to trace a batch beyond the first wholesaler, or failure to reach a simulated notification target – the manufacturer is required to implement corrective actions and repeat the mock recall within three months. The 2024 revision to the Recall and Rapid Alert System mandates that mock recall reports be uploaded to the National Drug Recall Portal, and the CDSCO can compare the mock recall performance with actual recall performance to assess the manufacturer’s truthfulness. Failure to conduct mock recalls or falsification of mock recall data is treated as a serious violation of Good Manufacturing Practices, leading to suspension of manufacturing licenses.
The U.S. Recall Procedure
Voluntary Initiation by Firm
In the United States, the Food and Drug Administration (FDA) strongly prefers that firms voluntarily initiate recalls whenever they discover or have reason to believe that a marketed drug product violates the Federal Food, Drug, and Cosmetic (FD&C) Act. Voluntary initiation is the most common pathway, accounting for over 90% of all drug recalls. When a firm – whether a manufacturer, repackager, or distributor – identifies a potential defect (e.g., out-of-specification test result, contamination, labeling error), it must immediately conduct an investigation and, if a violation is confirmed, notify the FDA’s appropriate district office within 24 hours. The firm is expected to submit a “recall strategy” outlining the depth of recall (wholesale, retail, or consumer level), the proposed public notification plan, effectiveness check procedures, and a timeline for completion. Unlike India, where the CDSCO plays a more directive role from the outset, the FDA allows the firm substantial discretion in designing the recall as long as the strategy is adequate to protect public health[58]. However, the firm must also submit a “Health Hazard Evaluation” (HHE) – a structured assessment of the risk posed by the violative product – which the FDA will review and may challenge. Voluntary initiation benefits the firm by demonstrating cooperation, which the FDA considers in subsequent enforcement decisions (e.g., whether to issue a warning letter, seek injunction, or pursue criminal charges). The firm may also conduct a “firm-initiated recall” even before the FDA has confirmed the violation, a practice known as “proactive recall,” which is viewed very favourably. The FDA’s role in voluntary recalls is primarily oversight: it monitors the recall’s progress, conducts its own HHE (which may differ from the firm’s classification), and has the authority to reclassify the recall or require additional actions[59].
FDA-Requested or Mandated Recalls
When a firm refuses or unreasonably delays initiating a voluntary recall for a product that the FDA believes violates the FD&C Act and poses a health hazard, the FDA can exercise its authority to request or mandate a recall. The legal basis for this authority is section 518(e) of the FD&C Act for devices and section 423 of the Food and Drug Administration Amendments Act (FDAAA) for drugs, though for most drugs the FDA relies on its general enforcement powers under sections 301 and 302. An “FDA-requested recall” is the softer option: the FDA sends a formal letter to the firm explaining the violation, the health hazard, and requesting that the firm initiate a recall. While phrased as a request, refusal is rarely advisable because the next step is a “mandated recall” under a court order. For a mandated recall, the FDA must first provide the firm with an opportunity for a hearing, then issue a final order requiring the recall[60]. If the firm still refuses, the FDA can seek a court injunction to compel the recall, and the firm may be held in contempt of court. In practice, FDA-mandated recalls are extremely rare – typically fewer than five per year – because most firms comply with a request. However, the mere existence of this authority gives the FDA significant leverage. Notably, the FDA does not have the statutory authority to unilaterally order a recall of all drugs without a court order; the exception is for infant formula, but not for pharmaceuticals. Therefore, the FDA’s power is indirect: it can seize products, suspend licenses, and recommend criminal prosecution, all of which are powerful incentives for voluntary compliance. The decision to escalate to a requested or mandated recall is made by the Office of Regulatory Affairs (ORA) in consultation with the relevant product center (e.g., CDER for drugs)[61].
Role of the Recalls and Shortages Branch
Within the FDA’s ORA, the Recalls and Shortages Branch (RSB) serves as the central coordinating unit for all recall activities involving FDA-regulated products, including drugs. The RSB is responsible for receiving recall notifications from firms, assigning a recall number, conducting the official Health Hazard Evaluation (HHE), assigning a recall classification (I, II, or III), and tracking the recall’s progress through completion. The RSB also maintains the public facing Recall Enterprise System (RES) and publishes the weekly FDA Enforcement Report, which lists all recalls classified within the previous two weeks. In addition to recall management, the RSB monitors drug shortages, because a recall of a medically necessary product can trigger or worsen a shortage. The RSB works closely with the Center for Drug Evaluation and Research’s (CDER) Drug Shortages Staff to assess whether a recall will impact patient access and to facilitate mitigation strategies, such as expediting approval of alternative suppliers or extending expiry dates for unaffected batches. The RSB also provides guidance to firms on recall strategies, including the appropriate depth of recall and the wording of public notifications. For complex recalls involving multiple products, batches, or supply chains, the RSB may convene a recall committee composed of experts from ORA, CDER, and the Office of the Chief Scientist to ensure consistent decision-making. The RSB’s role is largely administrative and technical, not enforcement-focused; enforcement actions resulting from a recall (e.g., warning letters, injunctions) are handled separately by the FDA’s Office of Compliance[62].
Recall Strategy and Health Hazard Evaluation (HHE)
Every recall initiated by a firm or requested by the FDA must include a written recall strategy and a Health Hazard Evaluation (HHE). The recall strategy specifies: the depth of recall (wholesale, retail, or consumer/user level), the date by which recall notifications will be issued, the method of notification (e.g., letter, email, phone call, press release), the instructions for consignees on how to respond (e.g., quarantine, return, destroy), the proposed effectiveness check plan (i.e., how the firm will verify that consignees have received and acted on the notification), and the timeline for recall completion. The FDA reviews the strategy and may require modifications if it deems the strategy inadequate[63]. The Health Hazard Evaluation (HHE) is a structured risk assessment conducted by the FDA (often using a standard HHE form) that considers six factors: (1) the nature and severity of the hazard (e.g., toxicity of contaminant, clinical impact of potency deviation); (2) the probability of occurrence of adverse health events given the defect; (3) the number of patients or products exposed; (4) the vulnerable population (e.g., children, elderly, pregnant women, immunocompromised); (5) the expected timeline for the hazard to manifest (immediate vs. delayed); and (6) the availability of alternative products or therapies. Based on the HHE, the FDA assigns the recall classification (I, II, or III). The firm may submit its own HHE, but the FDA’s classification is authoritative. The HHE also determines whether public notification is required (mandatory for Class I, discretionary for Class II, rarely for Class III) and whether the FDA will conduct its own audit of the recall effectiveness. The HHE is a living document that can be updated as new information emerges – for example, if additional adverse events are reported, a recall can be upgraded from Class II to Class I[64].
Public Notification and Enforcement Reports
The FDA places a strong emphasis on transparency in the recall process. For all Class I recalls and many Class II recalls, the FDA requires the firm to issue a public notification, typically a press release that is also posted on the FDA’s website, often within 24 to 48 hours of recall initiation. The press release must include the product name, lot numbers, NDC number, a photograph of the product if available, the nature of the defect, the health hazard posed, and instructions for consumers (e.g., “do not use, return to pharmacy for refund”). For Class I recalls, the FDA may also issue a nationwide alert through the FDA’s social media channels, MedWatch system, and direct emails to healthcare professional organizations. The FDA’s Enforcement Report is published weekly (usually on Wednesdays) and contains a complete listing of all recalls classified during the previous two weeks[65]. Each entry includes the recall number, firm name, product description, recall reason, recall classification, and the volume of product recalled. The Enforcement Report is searchable online and is used by healthcare systems, group purchasing organizations, and international regulators to track recall activity. Additionally, the FDA maintains a separate “Recall – Press Release” webpage for major recalls. Firms are also required to conduct their own notification to direct accounts; for consumer level recalls, this may include “Dear Doctor” letters, pharmacy bulletins, and even paid advertisements in major newspapers. The FDA does not charge firms for the cost of recall-related public notifications, but the firm bears all other recall costs (product return, destruction, notification, etc.). Failure to issue adequate public notification can lead to the FDA issuing its own public warning, which often carries more severe reputational consequences because it is perceived as an independent government warning.
Comparative Analysis: Initiation, Authority, and Enforcement
A pointwise comparative analysis of the Indian and U.S. recall procedures reveals important distinctions in initiation mechanisms, regulatory authority, and enforcement philosophies, while also highlighting areas of convergence driven by global harmonization efforts. First, regarding initiation of recalls, both systems rely primarily on voluntary action by the licensee or firm, reflecting the shared principle that manufacturers bear primary responsibility for product quality and safety. However, the Indian system places a relatively greater obligation on the licensee to self-classify and self-report within very short timelines (24 hours for Class I), whereas the U.S. system provides firms with slightly more flexibility to conduct an internal investigation before notifying the FDA, as long as the notification occurs “without unreasonable delay.” In practice, U.S. firms often notify the FDA within 24 to 48 hours, similar to India. A key difference is that Indian law explicitly criminalizes failure to voluntarily recall a dangerous product (Section 27), while U.S. law relies more on civil enforcement and the threat of FDA-requested recall rather than direct criminal penalties for non-recall. Second, concerning statutory authority to compel recalls, the CDSCO possesses broader direct powers than the FDA. The CDSCO can issue a unilateral recall order under Section 26A and physically seize product without a court order. In contrast, the FDA cannot unilaterally mandate a drug recall; it must either request a recall or seek a court order. This difference stems from the U.S. Constitution’s due process requirements and the FD&C Act’s specific language[66][67]. However, in practice, the FDA’s indirect powers (seizure, injunction, license suspension, criminal referral) are so potent that firms rarely refuse a recall request. The Indian approach is more paternalistic, reflecting a legal system where administrative authorities have greater latitude for unilateral action in public health emergencies. Third, regarding institutional infrastructure, the U.S. benefits from a dedicated Recalls and Shortages Branch with specialized expertise and a mature Recall Enterprise System, whereas India’s Recall and Rapid Alert System was only substantially upgraded in 2024 and is still being fully implemented across all states. The U.S. system’s integration with drug shortage monitoring is also more advanced; India’s current framework does not systematically link recalls to shortage risk assessment, though this is under development. Fourth, the depth-of-recall levels are conceptually similar (wholesale, retail, consumer) but the U.S. system uses more standardized definitions and has clearer guidance on when each level is appropriate, including detailed flowcharts. India’s levels are defined in general terms, leaving more discretion to the manufacturer and the CDSCO. Fifth, mock recalls are required in India only (since 2023 Schedule M), while the U.S. FDA recommends but does not mandate mock recalls for pharmaceutical manufacturers, except for certain device manufacturers under the Quality System Regulation. This is an interesting divergence: India has adopted a more prescriptive approach to preparedness testing, while the U.S. relies on routine inspections to verify distribution record accuracy. Sixth, public notification is more systematic and transparent in the U.S., with the weekly Enforcement Report providing a comprehensive public record of all classified recalls. India’s public notification remains more ad hoc; the CDSCO website lists some major recalls but not all, and there is no equivalent weekly report. The 2024 revision introduced a public dashboard, but its completeness and timeliness are still evolving. Seventh, enforcement consequences differ markedly. In India, failure to properly conduct a recall can lead to criminal prosecution, imprisonment, and license cancellation. In the U.S., criminal prosecution for recall-related violations is rare; the typical consequences are civil (warning letters, injunction, debarment from government contracts, reputational damage). The U.S. system relies more on market forces – a Class I recall of a major drug can wipe out hundreds of millions in revenue and trigger shareholder lawsuits – which often provides stronger incentives than criminal penalties. Eighth, international coordination is more formalized in the U.S., with established protocols for notifying foreign regulators through the International Recall Coordinating Group and mutual recognition agreements. India’s 2024 revision requires notification to importing countries for Class I recalls, but implementation remains uneven. Finally, both systems face common challenges: ensuring timely classification in the absence of complete data, preventing regulatory capture where large manufacturers receive lenient treatment, and managing the logistics of consumer-level recalls in fragmented distribution networks. The ongoing convergence – with India adopting risk-based HHE principles and the U.S. exploring mandatory recall authority for drugs – suggests that the two systems will continue to inform each other, driven by the global nature of pharmaceutical supply chains and the shared goal of protecting patient safety[68][69].
Future Directions and Recommendations
Strengthening India’s drug recall system requires a multi-pronged approach that addresses legal, administrative, technological, and clinical surveillance gaps. First and foremost, India should enact a standalone, mandatory recall law that explicitly defines the obligations of manufacturers, importers, and distributors, rather than relying on scattered provisions within the Drugs and Cosmetics Act and Rules. Currently, recall authority is implied through powers of suspension, seizure, and prohibition, but there is no dedicated legislation that compels a manufacturer to recall a defective product within a specified timeframe or that imposes graduated penalties for non-compliance. A standalone law would establish clear triggers for mandatory recall – such as a confirmed contamination, a serious adverse event cluster, or a foreign regulator’s safety alert – and would empower the Central Drugs Standard Control Organization (CDSCO) to issue binding recall orders without the need for prolonged litigation. The law should also mandate that all recalls, including Class III, be reported to a central repository, and should introduce criminal penalties for wilful concealment of defects while offering safe harbour provisions for manufacturers who voluntarily disclose and recall before any patient harm occurs. Second, enhancing harmonization between CDSCO and state regulators is critical because India’s federal structure divides authority: the CDSCO approves new drugs and oversees clinical trials, but state drug controllers license manufacturing facilities and conduct routine inspections. This fragmentation often leads to delayed or inconsistent recall actions – a state may classify a defect as Class II while another state treats it as Class I, or a recall ordered by CDSCO may not be effectively enforced by a state that has strained resources or different priorities. To address this, a unified recall protocol should be established through a legally binding memorandum of understanding between CDSCO and all state drug authorities, creating a joint recall coordination committee that meets monthly and shares a common database[70]. The CDSCO should also be granted the power to supersede state authorities in emergency recalls (Class I) and to directly deploy central inspectors when a state fails to act within 48 hours. Third, investment in digital traceability infrastructure is non-negotiable for a country that produces nearly one-fifth of the world’s generic medicines yet relies heavily on manual, paper-based distribution records. The current system often fails to locate the exact wholesale and retail points where a recalled batch was distributed, making consumer-level recalls nearly impossible. India should accelerate the implementation of a nationwide track-and-trace system based on barcode or RFID technology, building on the existing but incomplete requirement for unique product identifiers on drug packaging. This system should connect manufacturers, wholesalers, pharmacies, and hospitals to a central cloud-based platform that records every transaction in near real-time, enabling the CDSCO to identify precisely where a batch was shipped, how many units remain, and which patients may have received it. The government should provide subsidies or low-interest loans to small and medium-sized manufacturers to adopt this infrastructure, given their predominance in the Indian market. Fourth, strengthening pharmacovigilance integration with the recall system would transform recalls from reactive, product-focused actions into proactive, patient-centered interventions. India’s Pharmacovigilance Programme (PvPI) operates largely independently of the recall system, meaning that a signal of increased adverse events may not automatically trigger a recall investigation. A formal linkage should be created whereby any serious adverse event reported to the PvPI that is potentially product-related automatically generates a preliminary recall risk assessment. Furthermore, recall decisions should incorporate pharmacovigilance data to determine the depth of recall – for example, if a defect has already caused documented injuries, the recall must immediately escalate to consumer level, even if the defect’s inherent risk would otherwise suggest a retail-level recall. Integration would also enable post-recall surveillance to verify that removal of the defective product actually reduces adverse event rates[71][72].
Turning to the United States, while the FDA possesses a mature recall framework, several refinements could further enhance its effectiveness. Expanding unannounced international inspections is a priority, given that a large and growing proportion of prescription drugs, active pharmaceutical ingredients, and finished dosage forms consumed in the U.S. are manufactured abroad, particularly in India and China[73]. The FDA conducts foreign inspections but often announces them in advance, allowing facilities to temporarily correct deficiencies or hide records. Unannounced inspections – or inspections with minimal notice, such as 24 hours – would provide a truer picture of routine manufacturing conditions, including whether a facility has accurate distribution records necessary for rapid recall[74][75]. This requires legislative authorization and additional funding to maintain a standing corps of international inspectors who can travel on short notice, but the return on investment would be substantial, as seen in the wake of contaminated heparin and valsartan recalls that originated overseas. Second, enhancing real-time supply chain monitoring through electronic tracking of drug shipments from factory to pharmacy would dramatically reduce the time needed to identify recall depth and target notifications. While the Drug Supply Chain Security Act (DSCSA) has made progress in creating an interoperable electronic system for tracing prescription drugs, full implementation remains incomplete, and many small dispensers still lack the capability to receive and respond to recall alerts electronically. The FDA should mandate that all trading partners – manufacturers, repackagers, wholesale distributors, and dispensers – participate in a real-time recall alert system that pushes notifications directly to a pharmacy’s inventory management software, flagging recalled lots at the point of sale or dispensing. This would prevent the continued sale of recalled products that often occurs when recall notices are sent by postal mail and buried under paperwork. Third, leveraging Quality Management Maturity (QMM) programs would shift the regulatory paradigm from reactive recall oversight to proactive quality assurance[76][77]. QMM refers to a manufacturer’s ability to consistently produce products that meet specifications, continuously improve processes, and rapidly address deviations before they lead to widespread defects. The FDA has piloted QMM assessment frameworks, but these remain voluntary and are not systematically integrated into recall classification. Manufacturers with high QMM ratings could be granted reduced recall reporting burdens (e.g., monthly instead of weekly reports) or greater flexibility in designing their recall strategies, while those with low QMM ratings would face mandatory third-party audits and pre-approved recall plans that must be exercised immediately upon any defect detection. This creates a powerful market incentive for quality investment, as poor QMM would lead to higher regulatory scrutiny and potentially slower market access for new products[78][79].
Towards global convergence, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and the World Health Organization (WHO) play indispensable roles in aligning national recall systems. The ICH has produced guidelines on pharmacovigilance (E2 series) and quality risk management (Q9), but a dedicated ICH guideline on recall procedures and health hazard evaluation is conspicuously absent. Such a guideline would harmonize definitions of recall classes, standardize the elements of a health hazard evaluation (including quantitative risk thresholds), and establish mutual recognition of recall actions across ICH member countries. For example, if the FDA classifies a product as Class I based on a defect, the CDSCO and European Medicines Agency would accept that classification and automatically initiate their own recall procedures without re-evaluating the underlying data, provided the product was exported to their markets. The WHO, through its Global Surveillance System for substandard and falsified medical products, should create a real-time recall alert platform that all national regulators can access, with standardized reporting formats and translation into the six UN languages. This platform would be particularly valuable for low- and middle-income countries that lack their own sophisticated recall infrastructure and rely on alerts from larger regulators. Additionally, WHO could develop a benchmarking program for national recall systems, similar to its Global Benchmarking Tool for regulatory authorities, that assesses a country’s recall legal framework, inspection capacity, traceability systems, and pharmacovigilance integration. Countries that achieve a high benchmark score would gain expedited review status for product approvals in other benchmarked countries, creating a powerful incentive for regulatory strengthening. Finally, future research avenues should address the evidence gaps that currently limit recall system optimization. First, comparative effectiveness studies are needed to determine the optimal recall notification methods – for example, does a text message alert to a pharmacist achieve higher compliance than a letter or an email? Second, research on the economic impact of recalls on small manufacturers is critical, particularly in India, where a single recall can bankrupt a small firm, potentially incentivizing concealment. Studies should explore whether government-backed recall insurance pools or low-interest emergency loans could reduce this perverse incentive[80].
CONCLUSION
The comparative analysis of drug recall procedures in India and the United States reveals a complex landscape of convergent goals and divergent operational realities. Both regulatory systems share the fundamental objective of protecting public health by removing defective pharmaceutical products from the market swiftly and effectively. They have evolved from reactive, tragedy‑driven origins into increasingly proactive, risk‑based frameworks, recognizing that manufacturers bear primary responsibility for product quality and that voluntary recall is the preferred pathway. The three‑tier classification structure—Class I (most severe), Class II (moderate), and Class III (minor)—is common to both nations, as is the recognition that recall depth must align with the level of patient risk, ranging from wholesale to consumer level. However, important differences persist and carry practical implications. India’s CDSCO possesses broader unilateral statutory powers to compel recalls without court orders, reflecting a more paternalistic administrative tradition, yet its federal fragmentation with state regulators often leads to inconsistent classification and delayed enforcement. The U.S. FDA, while constitutionally constrained from unilaterally ordering drug recalls, has developed a more mature institutional infrastructure—the Recalls and Shortages Branch, the Recall Enterprise System, and the weekly Enforcement Report—that ensures greater transparency, standardized health hazard evaluations, and seamless integration with drug shortage monitoring. India’s 2023 Schedule M amendments and the 2024 revision of the Recall and Rapid Alert System represent significant strides toward harmonization, mandating mock recalls and creating a digital portal, but implementation remains uneven across states. The U.S. system’s reliance on civil enforcement and market forces (shareholder lawsuits, reputational damage) contrasts with India’s criminal penalties for non‑compliance, yet both face common challenges: regulatory capture, incomplete distribution records, and the logistical nightmare of consumer‑level recalls in fragmented supply chains. For policymakers and regulators, the study underscores the urgent need for India to enact a standalone, mandatory recall law that clarifies obligations, timelines, and graduated sanctions, while simultaneously resolving the central‑state coordination deficit through binding memoranda of understanding. Investment in digital traceability (barcode/RFID) and linkage with the Pharmacovigilance Programme of India (PvPI) would transform recalls from reactive clean‑ups to predictive, patient‑centred interventions. For the United States, despite its mature framework, refinements are warranted: expanding unannounced international inspections to address the reality of globalized supply chains, completing the Drug Supply Chain Security Act (DSCSA) to enable real‑time recall alerts at pharmacy points‑of‑sale, and integrating Quality Management Maturity (QMM) ratings into recall classification to incentivize proactive quality culture.
REFERENCES

Rohit Singh, Gulfsha Praveen, Comparative Analysis of Drug Approval Pathways in India v.s U.S.A, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 6536-6559, https://doi.org/10.5281/zenodo.20371759
 10.5281/zenodo.20371759
10.5281/zenodo.20371759