
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Guru Nanak College of Pharmaceutical Sciences, Dehradun.
This comprehensive review examines the current trends and future perspectives in pharmaceutical regulatory affairs (RA), tracing the evolution from a historical, reactive compliance function to a strategic, data-driven cornerstone of drug development. The article analyzes how scientific breakthroughs—including cell and gene therapies, mRNA platforms, and personalized medicine—are forcing a paradigm shift away from linear clinical development toward iterative, risk-based regulatory pathways. It explores the “digital-by-default” transformation driven by eCTD 4.0, end-to-end data traceability in GMP and CMC, and the integration of artificial intelligence (AI) for submission automation, predictive compliance, and regulatory intelligence. The review assesses global harmonization efforts, highlighting collaborative initiatives such as Project Orbis and the Access Consortium, while acknowledging persistent disparities in approval timelines across regions. Patient-centric trends are examined through the growing regulatory acceptance of real-world evidence (RWE), patient-focused drug development, and transparency mandates. Accelerated pathways for advanced therapies—including RMAT, PRIME, and SAKIGAKE—are discussed alongside pending EU legislative reforms. Finally, the article addresses supply chain resilience, intensified FDA inspections in China and India, and the imperative of upskilling regulatory professionals with data science and AI competencies. The future horizon envisions routine collaborative assessments, AI as a strategic engine, and pervasive data-driven oversight by agencies like the FDA and EMA. The conclusion asserts that regulatory affairs has become an indispensable strategic partner in delivering safe, effective, and accessible therapies to patients worldwide.
The role of Regulatory Affairs (RA) within the pharmaceutical industry has undergone a profound metamorphosis, transitioning from a back-office, administrative function to a strategic cornerstone of product development and corporate governance. For decades, the regulatory professional was often viewed as a necessary but obstructive gatekeeper—a custodian of compliance tasked with compiling dossiers, managing submissions, and ensuring that marketing authorization applications (MAAs) strictly adhered to a fixed set of guidelines[1]. This historical paradigm, rooted in a linear, post-hoc approach to regulation, was largely reactive. The primary mandate was to avoid regulatory rejection by checking boxes and rectifying deviations only after they had occurred during clinical development or manufacturing[2]. However, this traditional model, while functional for the blockbuster era of the late 20th century, is being rendered obsolete by a confluence of scientific, technological, and societal forces. Today, the industry finds itself in the grip of an unprecedented transformation, driven by advanced therapies, digital health technologies, real-world evidence (RWE), and a global push for accelerated patient access[3][4][5]. Consequently, the modern Regulatory Affairs professional has been propelled to the forefront of the C-suite, evolving into a strategic architect who shapes clinical development plans, engages in continuous scientific dialogue with health authorities, and navigates a fragmented yet interconnected global regulatory landscape. Understanding this evolution requires not merely a historical comparison but a recognition that the pace and complexity of change today are fundamentally different in kind, not just in degree, from any previous era[6][7].To appreciate the magnitude of today’s shift, one must first acknowledge the historical milestones that built the modern regulatory framework, as each previous upheaval added a layer of complexity to the RA role. The modern era of drug regulation is often traced to the Elixir Sulfanilamide tragedy of 1937 in the United States, which led to the 1938 Federal Food, Drug, and Cosmetic (FD&C) Act, mandating proof of safety before marketing. The RA function born from this was rudimentary, focused on collating safety data for submission[8][9]. The next seismic shift was the Thalidomide disaster of the early 1960s, which catalyzed the 1962 Kefauver-Harris Amendments[10][11]. These amendments revolutionized the industry by demanding not just safety, but substantial evidence of efficacy from adequate and well-controlled clinical trials. This legislation gave birth to the modern Investigational New Drug (IND) application process and formalized the role of the RA department in designing trial protocols that would satisfy regulatory scrutiny[12][13]. For decades following, the RA role was defined by a command-and-control model. Health authorities like the FDA and the European Medicines Agency (EMA) issued prescriptive guidelines, and the RA’s job was to ensure absolute conformity. The 1980s and 1990s brought the Hatch-Waxman Act (creating generics) and the advent of the International Council for Harmonisation (ICH), which helped standardize technical requirements across the US, EU, and Japan. While these were significant shifts, they primarily refined existing processes; the RA professional remained a specialist in documentation, submission formatting (from paper to eCTD), and post-market compliance. The core modus operandi was linear: develop, test, submit, approve, and monitor[14][15].
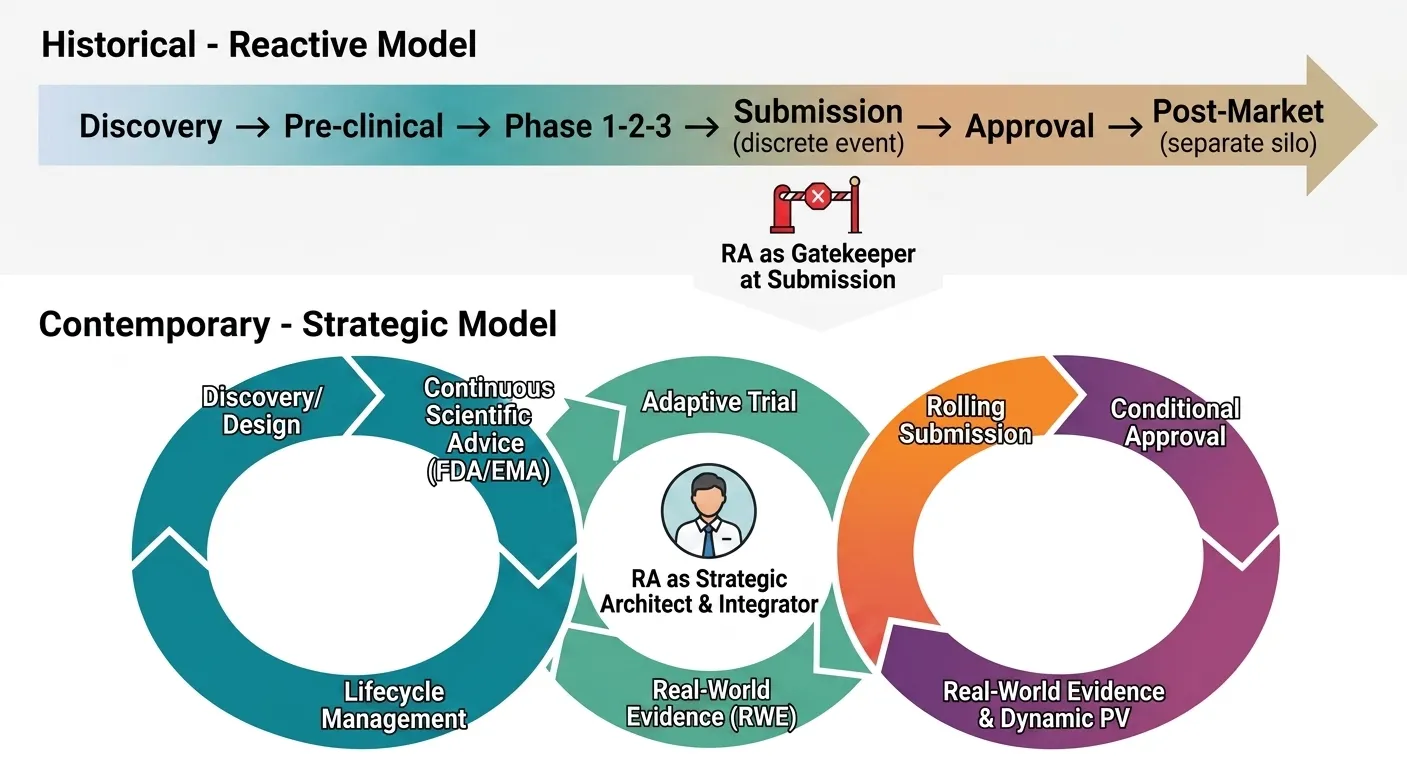
Fig: 1 Evolution of the Regulatory Affairs Paradigm
The contemporary transformation, however, shatters this linear model entirely. The first driver of this change is the scientific revolution, specifically the rise of advanced therapy medicinal products (ATMPs) including cell and gene therapies, mRNA platforms, and personalized medicine[16][17]. Unlike the small molecules and monoclonal antibodies of the past, these therapies are inherently variable, patient-specific, and often curative after a single administration[18][19]. This demands a radically different regulatory approach. There are no long-term historical precedents for gene editing; traditional phase 1-2-3 trial designs are often infeasible or unethical. Regulatory frameworks, such as the FDA’s RMAT (Regenerative Medicine Advanced Therapy) designation and EMA’s PRIME scheme, have adapted by promoting iterative, real-time scientific advice. Consequently, the RA professional is no longer a passive recipient of guidelines but an active co-creator of novel regulatory pathways. They must engage in continuous, risk-based assessments with agencies, negotiate surrogate endpoints, and design long-term follow-up surveillance for durable effects that may manifest years later. This requires a level of scientific fluency and strategic foresight unheard of in the 1990s. The RA expert must now debate the intricacies of vector shedding in AAV gene therapies or the immunogenicity risks of mRNA lipids, shifting the role from a legal-compliant one to a deeply scientific, translational one[19][20]. The digital transformation of healthcare has thrust RA into the unfamiliar territory of software, algorithms, and data integrity. The proliferation of digital health technologies (DHTs)—from wearable sensors that measure vital signs in decentralized trials to Software as a Medical Device (SaMD) that uses AI to diagnose conditions—blurs the line between drug and device. A pharmaceutical company developing a digital therapeutic for diabetes or a companion diagnostic for cancer immunotherapy must now navigate a regulatory ecosystem historically siloed from drug regulation. Furthermore, the use of real-world data (RWD) from electronic health records, insurance claims, and patient registries is no longer a fringe concept but a central pillar of regulatory science. The FDA’s framework for using RWE to support label expansions or post-approval requirements represents a paradigm shift from relying solely on explanatory randomized controlled trials (RCTs). For RA, this means mastering data governance, statistical methodologies for causal inference, and the validation of algorithms. They must answer novel questions: How does a health authority verify the integrity of RWD collected outside a controlled trial[23]. What constitutes a “fit-for-purpose” dataset for a supplemental application[21][22]. This new terrain demands that RA professionals possess data literacy and an understanding of artificial intelligence, a far cry from the document management skills of a previous generation[24][25]. The role is now predictive and analytical, tasked with mining existing data for regulatory opportunities before a formal submission is even drafted. The global regulatory landscape has fragmented while simultaneously becoming more interdependent, creating a complex web that defies the old model of “harmonization.” While ICH has made strides, the last decade has witnessed the rise of powerful, independent regulatory authorities in emerging markets like China (NMPA), Brazil (ANVISA), and India (DCGI). These agencies no longer simply defer to the FDA or EMA; they demand local clinical data, distinct labeling, and even unique manufacturing standards. The geopolitical decoupling, accelerated by the COVID-19 pandemic and supply chain vulnerabilities, has added another layer of complexity. RA must now navigate not only scientific dossiers but also trade regulations, data localization laws (e.g., GDPR in Europe, PIPL in China), and national security concerns regarding drug ingredient sourcing. Simultaneously, the success of initiatives like Project Orbis (which facilitates concurrent submission and review of oncology drugs among multiple international partners) demonstrates a new model of collaborative, parallel review[26][27]. This is a double-edged sword: it speeds access but requires a harmonized submission strategy developed in real-time. The modern RA professional must be a geopolitical strategist, capable of orchestrating a global submission plan that accounts for divergent requirements, rolling reviews, and varying degrees of digital maturity across dozens of markets[28][29]. They lead not through a static global playbook but through agile, territory-specific engagement that anticipates regulatory divergence as a feature, not a bug. Perhaps the most defining feature of today’s unprecedented transformation is the move from a reactive, submission-based mentality to a proactive, lifecycle-based continuum of engagement. Regulatory authorities themselves have changed their posture. The FDA’s “breakthrough therapy” designation, EMA’s adaptive pathways, and Japan’s SAKIGAKE designation system embody a regulatory science that is built for speed and iteration. These pathways encourage early and frequent interaction—sometimes as early as phase 1—to align on clinical trial designs, chemistry and manufacturing controls, and patient safety monitoring[30][31]. For RA, this means the “submission” is no longer a discrete event but a rolling, living process. They are now embedded in every stage of product development, from lead optimization (advising on the regulatory implications of a molecule’s physicochemical properties) to commercialization and lifecycle management. This shift is most evident in the post-marketing environment, where risk management plans (RMPs) have evolved into dynamic, data-driven surveillance systems. The RA professional oversees not just periodic safety update reports but digital dashboards that track product performance in real-time, triggering rapid regulatory responses. This agency-like mindset within a pharmaceutical company is a radical departure from the past, where regulation was something that happened to the product at the end of the pipeline[32][35].
The COVID-19 pandemic served as an extreme stress test that permanently accelerated this transformation. The development and authorization of multiple mRNA vaccines in under a year compressed a decade’s worth of regulatory interaction into months. RA teams had to negotiate manufacturing changes in real-time as scale-up occurred in parallel with clinical trials. They managed global supply chains where an export ban in one country could jeopardize a filing in another[36][37]. They worked with health authorities to implement platform technologies and master protocols, establishing new precedents for how vaccines and therapeutics for future pandemics will be regulated. The pandemic also normalized the use of virtual inspections and remote regulatory audits, destroying the long-held assumption that in-person presence was non-negotiable[38][39]. It forced the adoption of electronic common technical document (eCTD) structures for rapid, rolling submissions and demonstrated that decentralized clinical trials could generate valid evidence. The RA professional emerged from the pandemic not as a bureaucratic necessity but as a crisis manager and strategic enabler, having proven that barriers could be dismantled when patient need was urgent. This legacy has permanently raised the bar for speed and agility across all therapeutic areas, not just infectious diseases[40].
In comparing historical shifts to today’s transformation, a clear qualitative difference emerges. Historical shifts—safety, then efficacy, then harmonization—were about adding new, discrete requirements to a stable, linear process. The solution was more documentation, more specialization, and more checklists. The RA role expanded in scope but remained fundamentally administrative and reactive. Today’s transformation, by contrast, is about liquefying the process itself. The boundaries between development and review, between pre-market and post-market, and between drug, device, and data have dissolved[41][42]. The RA professional must be a polymath: part scientist to understand gene editing, part data analyst to harness RWE, part diplomat to navigate geopolitical tensions, and part futurist to anticipate AI-driven adaptive trials. This is not simply a job enlargement; it is a job redefinition. The risk of failure has also transformed. In the past, regulatory failure meant a complete response letter and a delay. Today, in the era of accelerated approvals and conditional marketing authorizations, regulatory failure might mean a product is approved but then withdrawn due to post-market safety signals, causing catastrophic reputational and financial damage. Thus, the RA’s role in designing robust, continuous pharmacovigilance and risk-minimization strategies is as critical as the initial approval itself[43][44].
Furthermore, the strategic integration of RA with commercial and medical affairs functions is now non-negotiable. In the historical model, RA handed the approved label to commercial and walked away. Today, the label is a living document, updated with RWE, comparator studies, and patient-reported outcomes. The RA professional works alongside market access teams to ensure that the evidence package satisfies not just regulators but also health technology assessment (HTA) bodies like NICE in the UK or IQWiG in Germany. The increasing alignment of regulatory and HTA evidence requirements, through initiatives like parallel scientific advice, means RA is now a critical partner in demonstrating value, not just safety and efficacy. This elevates the function from a cost center to a strategic driver of commercial success. A drug may be regulatory-approved, but without a robust strategy for demonstrating comparative effectiveness and real-world value, it will not be reimbursed. The modern RA team is instrumental in designing the clinical program to satisfy both masters simultaneously[45][46].
Education and competency frameworks for RA professionals are finally catching up to this new reality. The traditional career path of a regulatory writer or submissions specialist is being augmented by requirements for advanced degrees in data science, molecular biology, or public health. Soft skills—negotiation, risk communication, and cross-functional leadership—are now as important as technical knowledge of 21 CFR or EudraLex. Professional organizations like the Regulatory Affairs Professionals Society (RAPS) have overhauled their certifications to emphasize strategic thinking, global intelligence, and lifecycle management. The modern RA leader is a member of the product development team from day zero, often serving as the project manager who integrates clinical, non-clinical, and CMC (Chemistry, Manufacturing, and Controls) inputs into a coherent regulatory narrative[49][50]. They are the ones who identify potential regulatory hurdles in a novel CAR-T construct long before a single patient is dosed, or who see an opportunity for an orphan designation in a rare disease subset that opens the door to premium pricing and market exclusivity[51].
The New Regulatory Imperative: Digitalization, Artificial Intelligence, Patient Centricity, and Supply Chain Resilience in Modern Pharmaceuticals
The pharmaceutical regulatory landscape is undergoing a structural revolution driven by the convergence of digital technologies, artificial intelligence (AI), evolving patient expectations, and geopolitical realignments[52]. Gone are the days when regulatory affairs meant static submissions and retrospective compliance checks. Today, regulators and industry alike are embracing a “digital-by-default” paradigm, integrating end-to-end data traceability, leveraging AI for oversight and workflow automation, pursuing global harmonization through reliance models, embedding patient-centricity via real-world evidence (RWE), designing agile pathways for advanced therapies, and fortifying supply chain resilience against global shocks. This multifaceted transformation is not merely a series of incremental upgrades but a fundamental re-engineering of how medicinal products are developed, reviewed, approved, and monitored across their entire lifecycle[53].
At the heart of this transformation lies the digital-by-default paradigm, championed by leading health authorities such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Both agencies have launched ambitious initiatives to replace paper-based, fragmented processes with seamless, electronic, data-driven ecosystems.[54] The FDA’s Technology Modernization Action Plan (TMAP) and the EMA’s Digital Business Transformation strategy aim to create a regulatory environment where submissions, reviews, and inspections are conducted using structured, machine-readable data from the outset. The cornerstone of this initiative is the mandatory use of the electronic Common Technical Document (eCTD) and its evolution toward eCTD 4.0. Unlike its predecessor (eCTD 3.2.2), which still relied on PDF-based unstructured content, eCTD 4.0 is built on a fully relational, hierarchical data model using HL7 FHIR (Fast Healthcare Interoperability Resources) and standardized XML schemas. This allows individual data elements—such as a single adverse event or a manufacturing process parameter—to be tagged, queried, and analyzed across submissions without manual extraction[55]. For regulatory affairs professionals, eCTD 4.0 enables a shift from document management to data management, where submission quality is measured by data conformance and completeness rather than formatting correctness. The FDA’s pilot programs for eCTD 4.0 and the EMA’s SPOR (Substances, Products, Organisations, Referentials) data management system exemplify this push toward a unified, authoritative data backbone. This digital paradigm extends deeply into Good Manufacturing Practice (GMP), Chemistry, Manufacturing, and Controls (CMC), and quality systems through the demand for end-to-end data traceability and audit readiness. Regulators now expect that every quality attribute, every batch record, and every environmental monitoring reading is captured in an electronic system that provides an immutable, time-stamped audit trail[56][57]. The concept of “audit readiness” has evolved from preparing binders of paper records to maintaining continuous, real-time access to manufacturing execution systems (MES), laboratory information management systems (LIMS), and electronic batch records (EBR). The FDA’s focus on data integrity, codified in guidance such as “Data Integrity and Compliance With Drug CGMP,” emphasizes the ALCOA+ principles (Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, Available). In practice, this means that every CMC change—whether a new supplier of an excipient or a modification in a lyophilization cycle—must be traceable through change control systems that feed directly into the regulatory submission. For global companies, the challenge is harmonizing disparate manufacturing sites onto a single digital quality management system (QMS) that allows inspectors to remotely query data. The COVID-19 pandemic accelerated the use of virtual and hybrid inspections, where regulators request live access to digital records without stepping foot on site. Consequently, organizations that have not yet digitized their GMP documentation face not only compliance risks but also significant delays in marketing authorization and supply continuity.
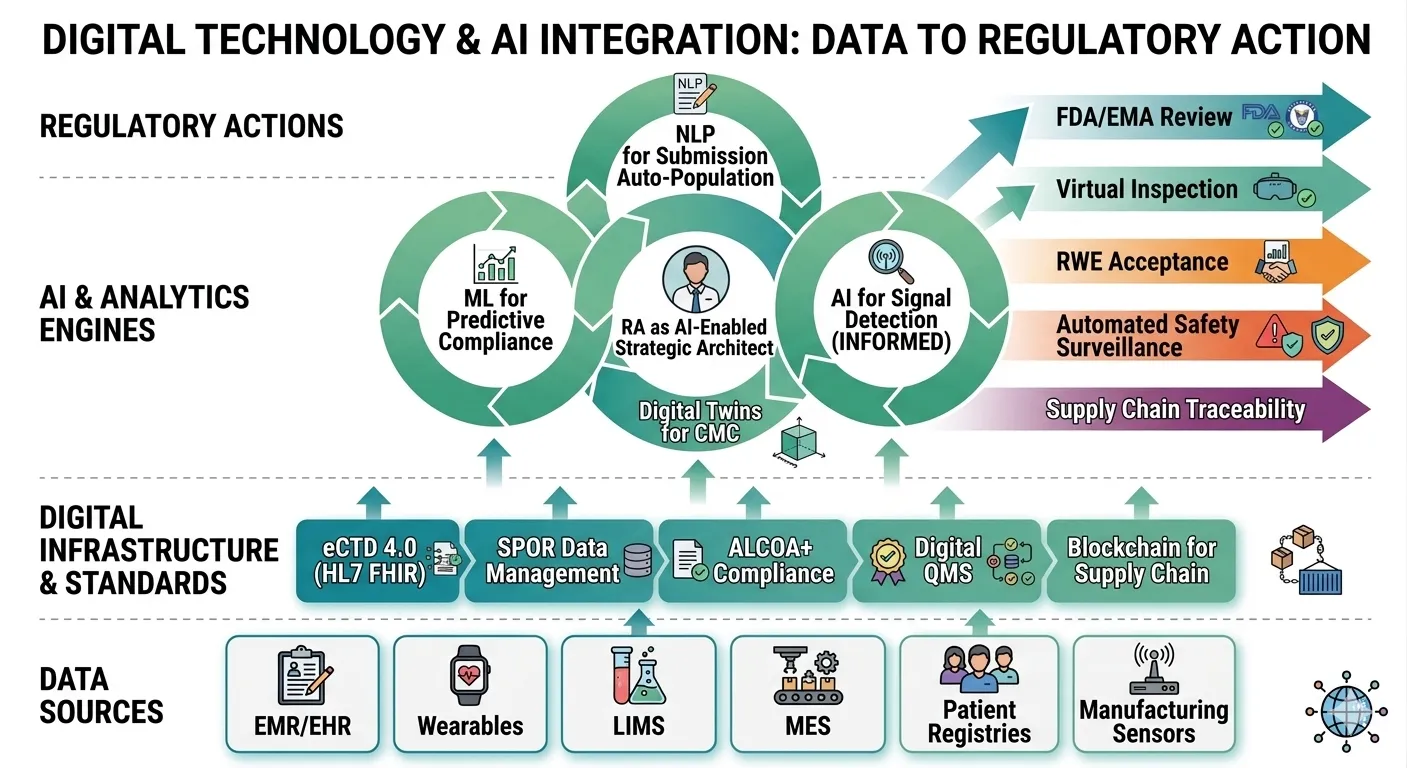
Fig: 2 Digital technologies and AI are integrated from data source to regulatory action
The integration of artificial intelligence (AI) into regulatory oversight represents the next frontier. Both the FDA and EMA are actively exploring and deploying AI tools for facility reviews, product evaluation, and pharmacovigilance[59]. For example, the FDA’s Information Exchange and Data Transformation (INFORMED) project uses machine learning to triage thousands of adverse event reports, identifying safety signals more rapidly than manual review. Similarly, the EMA’s AI pilot program tests algorithms for assessing variations to marketing authorizations, automatically checking completeness of CMC dossiers against predefined rules. On the industry side, AI is automating regulatory workflows with profound impact on submission preparation and quality assurance. Natural language processing (NLP) tools can parse clinical study reports, extract critical efficacy and safety tables, and populate eCTD modules with minimal human intervention. Machine learning models are being trained to predict regulatory questions based on historical review data, allowing teams to proactively address potential deficiencies. AI-driven quality assurance systems can scan thousands of pages of regulatory submissions for formatting errors, missing hyperlinks, or inconsistent terminology in seconds—a task that once took weeks[60][61]. However, the deployment of AI in regulatory affairs raises novel challenges around data governance and talent. Building an AI-ready talent pool requires regulatory professionals who understand not only the law and science but also data architecture, algorithm validation, and bias mitigation. Companies are creating hybrid roles such as “regulatory data scientists” and investing in upskilling existing staff in Python, SQL, and AI ethics. A robust data governance framework—covering data provenance, version control, and access permissions—is a prerequisite for any AI initiative; garbage in, garbage out remains the immutable law of machine learning[62].
Parallel to the digital and AI revolutions, global regulatory harmonization and convergence have taken on new urgency and new forms. The historic model of sequential national submissions is being replaced by work-sharing, reliance, and collaborative assessment initiatives. The FDA’s Project Orbis, which enables concurrent review of oncology products among multiple international partners (Australia, Canada, Switzerland, UK, and others), has become a template for other therapeutic areas. The EMA’s ACCESS consortium and the African Medicines Agency (AMA) are building frameworks where a stringent regulatory authority’s approval can be relied upon by countries with limited review capacity[63][64]. The Access Consortium, comprising Australia, Canada, Singapore, Switzerland, and the UK, has operationalized a “single assessment” process for new chemical entities, where one lead regulator performs the scientific review and others rely on it while adding national-specific labeling or risk management requirements. These models reduce duplication, shorten time to patient access, and conserve regulatory resources. The push for unified submission standards remains anchored in ICH (International Council for Harmonisation) guidelines, particularly the Common Technical Document (CTD) and eCTD. ICH M4 and M8 have created a globally accepted structure for dossiers, but implementation gaps persist—Russia, Brazil, and China have local variations that still require reformatting. Looking forward, the next horizon is routine collaborative assessments where a single submission package is reviewed simultaneously by a “regulatory college” representing multiple jurisdictions. The success of the COVID-19 joint assessment by the EMA and the World Health Organization (WHO) has provided a proof of concept. To achieve this at scale, regulators will need to align on data standards, acceptance criteria, and confidentiality arrangements—a challenge that is as political as it is technical. A defining trend reshaping regulatory practice is patient-centricity, moving from a paternalistic model to one where patient perspectives directly inform benefit-risk decisions. The most significant manifestation is the regulatory acceptance of real-world evidence (RWE) for decision-making. The FDA’s RWE Framework, established under the 21st Century Cures Act, outlines how data from electronic health records, claims databases, patient registries, and even wearable devices can support new indications, post-marketing requirements, or even approval of rare disease drugs when randomized trials are infeasible[66]. The EMA’s DARWIN EU project builds a coordinated network of real-world data sources to generate evidence for regulatory use. Notable case studies include the FDA’s approval of palbociclib for certain breast cancer patients based on real-world overall survival data from the Flatiron Health database, and the EMA’s use of registry data to extend the indication of a CAR-T therapy. Patient-centered medical product development goes further, involving patients in trial design, endpoint selection, and benefit-risk frameworks[67][68]. The FDA’s Patient-Focused Drug Development (PFDD) program and the EMA’s “Patients’ and Consumers’ Working Party” routinely collect patient experience data that can qualify as substantial evidence for a labeling claim. For instance, input from patients with spinal muscular atrophy (SMA) influenced the design of trials for gene therapy Zolgensma, prioritizing motor milestone achievement as a clinically meaningful endpoint. The impact of transparency and public trust cannot be overstated. Regulators now publish detailed review summaries, Clinical Study Reports (CSRs), and even raw data (anonymized) to foster accountability. The EMA’s Clinical Data Publication policy, which releases CSR redacted versions, has increased both public scrutiny and trust. However, it also demands that RA professionals become adept at managing data anonymization and protecting commercial confidential information while embracing openness[69][70].
The emergence of accelerated pathways and agile regulation for advanced therapies represents perhaps the most radical departure from traditional regulatory frameworks. Cell and gene therapies, mRNA platforms, and cancer vaccines defy the conventional phase 1-2-3 development paradigm. A single dose of a gene therapy may be curative, making large, long-term randomized trials ethically and practically challenging. Regulators have responded with innovations in accelerated approval pathways. The FDA’s Regenerative Medicine Advanced Therapy (RMAT) designation, EMA’s PRIME (PRIority MEdicines) scheme, and Japan’s SAKIGAKE (early access) all offer early and frequent scientific advice, rolling reviews, and potential approval based on surrogate or intermediate endpoints. The landmark approval of the first CRISPR-based gene therapy (Casgevy for sickle cell disease) involved a single-arm trial with follow-up for a limited cohort, relying on biomarker data (fetal hemoglobin levels) as a surrogate for clinical benefit. Similarly, the mRNA platform technology—which proved its worth in COVID-19 vaccines—is now being regulated under a platform approach, where master files for the lipid nanoparticle delivery system can be leveraged across multiple product candidates. Single-trial approvals, once an exception, are becoming more common for rare diseases and breakthrough therapies. The FDA’s accelerated approval of aducanumab for Alzheimer’s disease based on a surrogate endpoint (amyloid beta reduction) sparked intense debate but also demonstrated the flexibility—and controversy—of modern regulatory agility. Looking to the future, the EU General Pharmaceutical Legislation is undergoing its most significant revision in two decades. The proposed reforms aim to reduce the standard review time, introduce “regulatory sandboxes” for novel technologies, and create a “cradle-to-grave” regulatory science support for advanced therapies. Key proposals include streamlining orphan designation, reducing exclusivity periods for products that do not address unmet needs (to incentivize true innovation), and mandating that all new drug applications include a plan for patient engagement. Critics worry that the EU reforms may overly bureaucratize agility, while proponents argue they will harmonize the fragmented national procedures that currently delay patient access across member states. Regardless, the direction is clear: regulation must adapt to the pace of science, not the other way around.
Future Perspectives and Emerging Challenges
As the pharmaceutical industry looks toward the next decade, the regulatory affairs function stands at a precipice of profound change. The foundations laid by digitalization, artificial intelligence, patient centricity, and accelerated pathways are now converging into a new strategic reality. The future of regulatory affairs will be defined not by incremental improvements but by a fundamental reimagining of how global regulators collaborate, how technology transforms compliance from reactive to predictive, how leading agencies evolve their oversight models, and how regulatory professionals themselves must upskill to remain relevant. This final horizon demands that organizations move beyond adapting to change and instead become architects of the new regulatory order. The next horizon for regulatory harmonization is no longer about merely aligning technical requirements but about operationalizing collaborative assessments at scale. The past decade’s work-sharing initiatives Project Orbis, the Access Consortium, and the EMA’s reliance-based procedures have proven that concurrent or joint reviews are feasible. However, these remain largely confined to oncology, orphan drugs, or a handful of well-aligned jurisdictions. The future requires strengthening international collaborative assessments to cover mainstream therapeutics and diverse regulatory ecosystems. The International Council for Harmonisation (ICH) has recently launched a reflection paper on “Global Regulatory Cooperation” that envisions a framework where a single application package, prepared once, could be reviewed simultaneously by multiple authorities using a shared assessment report. This would reduce redundant questions, cut approval timelines by months, and free up constrained regulatory resources. Yet, significant barriers remain. Legal frameworks differ: some authorities cannot share confidential commercial information without bilateral treaties. Review cultures diverge: the FDA’s statistical rigor may conflict with the EMA’s more holistic benefit-risk balancing. And resource asymmetries persist: a well-staffed authority like the Japanese PMDA can conduct deep reviews, while smaller agencies may lack the expertise to contribute meaningfully.
CONCLUSION
The trajectory of pharmaceutical regulatory affairs over the past century represents one of the most profound professional transformations in modern industry. From its origins as a document-centric, reactive gatekeeping function born of tragic drug disasters, RA has ascended to become a strategic architect of product development, a guardian of continuous patient safety, and a driver of global public health. The evidence presented throughout this review is unequivocal: the old paradigm of linear, submission-based compliance is no longer fit for purpose. Today’s regulatory environment demands agility, scientific fluency, data literacy, and geopolitical awareness. The convergence of digital technologies, artificial intelligence, patient-centric values, and novel therapeutic modalities has liquefied traditional boundaries between development and review, pre-market and post-market, drug and device, local and global. Looking forward, the next decade will be defined by whether the pharmaceutical industry and regulatory agencies can collectively operationalize the promise of collaborative assessments, moving from isolated work-sharing pilots to routine, simultaneous multi-jurisdictional reviews. The evolution of AI from a productivity tool to a strategic engine will hinge on robust data governance and the emergence of new competencies in model assurance and predictive compliance. The FDA’s vision of pervasive, data-driven oversight—where real-time manufacturing data streams inform continuous regulatory monitoring—will demand that companies embed compliance into every digital record and algorithm. Meanwhile, the EMA’s proposed legislative overhaul and the maturation of agencies in emerging markets will test the resilience of global harmonization. Crucially, the human element of regulatory affairs cannot be overlooked. The skills gap between traditional document specialists and tomorrow’s data-savvy strategists is widening. Organizations must invest urgently in upskilling—creating hybrid roles, fostering digital maturity, and cultivating a culture where regulatory professionals are empowered to interpret AI outputs, negotiate with authority, and lead cross-functional teams. The regulatory professional of 2030 will not be measured by the volume of submissions filed but by the strategic value added to product development, the speed and certainty of patient access achieved, and the integrity of safety systems maintained over the entire product lifecycle.
REFERENCES

Sahil Kumar, Chetan Singh, Current Trends and Perspectives in Pharmaceutical Regulatory Affairs, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 6589-6602, https://doi.org/10.5281/zenodo.20371862
 10.5281/zenodo.20371862
10.5281/zenodo.20371862