
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Regulatory Affairs, Chalapathi Institute of Pharmaceutical Sciences, Guntur, Andhra Pradesh, India 522034
This comparative study analyzes the regulatory frameworks that oversee medical devices in the United States and Japan, highlighting the processes, challenges, and future directions. The U.S. FDA and Japan's PMDA regulate these devices with distinct classification systems and approval pathways tailored to their respective healthcare landscapes. The FDA employs a risk-based approach with 510(k), Premarket Approval (PMA), and De Novo processes, while Japan emphasizes local clinical trials for higher-risk devices under PMDA and Ministry of Health, Labour, and Welfare oversight. Key differences include regulatory timelines, fee structures, and post-market surveillance systems. Challenges such as high approval costs, regulatory complexity, and harmonization issues affect market entry and innovation. Future directions suggest enhanced global collaboration, streamlined approval processes, and robust post-market surveillance to support innovation while ensuring public health. This review says the statistical approach of medical device applications and number of applications approved and rejected in USA and Japan.
The global medical device industry plays a vital role in advancing healthcare technology and improving patient outcomes, with the USA and Japan standing as one of the largest and most impactful markets globally. These nations have established sophisticated regulatory frameworks through their respective authorities like the FDA and PMDA responsibility is to ensure the Safety, efficacy, and quality of medical devices while fostering innovation. The regulatory frameworks in these countries showcase their unique approaches to overseeing medical devices.The FDA employs a three-tiered classification system with specific approval pathways including 510(k), Premarket Approval (PMA), and De Novo processes, tailored to different risk levels. In contrast, Japan's PMDA operates under a four-tier classification system, with a particular emphasis on local clinical trials for higher-risk devices. Both systems demonstrate unique characteristics in their approval processes, fee structures, and post-market surveillance requirements, which significantly impact manufacturers' strategies for market entry. Recent data from 2022-2024 reveals interesting patterns in approval rates and regulatory decisions. While the U.S. sees a high volume of 510(k) approvals annually (over 1,700), Japan shows steady growth in Class II device approvals (exceeding 2,000 per year). These statistics underscore the regulatory rigor of each system and the dynamic evolution of the medical device industry in both markets. A comprehensive understanding of these regulatory frameworks and their practical implications is crucial for stakeholders involved in the design, manufacturing, and distribution of medical devices in these prominent healthcare sectors. The years 2022 to 2024 have provided valuable insights into these regulatory systems. The U.S. has demonstrated a consistently high volume of approvals, particularly under the 510(k) process, indicative of its streamlined and efficient approach to moderate-risk devices. Meanwhile, Japan has showcased steady growth in Class II device approvals, reflecting its systematic and safety-oriented methodology. Both systems play critical roles in shaping market strategies for manufacturers, who must navigate these frameworks to introduce innovative solutions effectively.
Table:1 Various Regulatory Authorities Worldwide
|
Country/Region |
Authority |
Year |
|
United States |
FDA- Food and Drug Administration |
1906 |
|
Europe |
EMA- European Medicine Agency |
1995 |
|
Japan |
PMDA- Pharmaceuticals and Medical Devices Agency |
2004 |
|
Canada |
Health Canada |
1993 |
|
United Kingdom |
MHRA-Medicines and Healthcare Products Regulatory Agency |
2003 |
|
Australia |
TGA- Therapeutic Goods Administration |
1989 |
DEFINITION:
A "medical device" is any instrument, apparatus, implement, machine, appliance, implant, in vitro reagent, software, substance, or similar object that the producer intends to employ, either alone or in combination, for specific medical purposes involving humans.
JAPAN:
Regulatory body: PMDA- Pharmaceuticals and Medical Devices Agency
The medical device industry plays a vital role in advancing healthcare and improving patient outcomes. To ensure the safety, efficacy, and quality of these devices, nations worldwide have implemented regulatory frameworks to oversee their registration and approval processes. As a leading global market for medical devices, Japan has established a robust regulatory system designed to protect public health while encouraging innovation in healthcare technologies. The Pharmaceuticals and Medical Devices Agency (PMDA), under the Ministry of Health, Labour, and Welfare (MHLW), oversees the regulation of medical devices in Japan, ensuring they comply with stringent safety standards before market approval.
Table:2 Medical devices classification according to PMDA
|
Class |
Description |
Examples |
|
Class- I |
Extremely low-risk |
X-ray film, steel surgical instruments |
|
Class- II |
Low- risk |
MRI units, dental alloys |
|
Class- III |
Medium- risk |
Artificial bones, Dialyze |
|
Class- IV |
High- risk |
Pacemaker, Artificial heart valves |

Figure:1 The process of medical devices registration in Japan
The process for medical device approval in Japan involves several structured steps:
Table:3 Fee Structure by Device Class Japan:
|
Device Class |
Registration Fee (?) |
|
Class I |
No fee |
|
Class II |
$86,000 |
|
Class III |
$86,000 |
|
Class IV |
$147,000 |
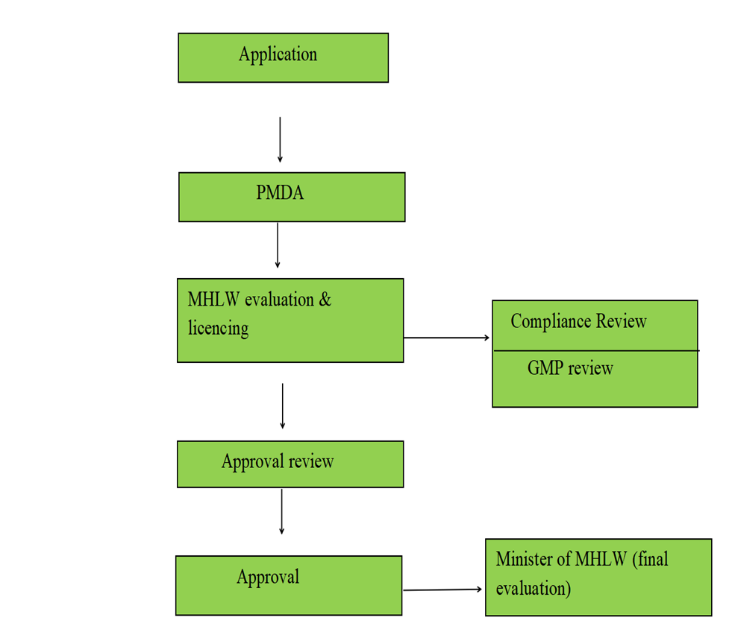
Figure:2 Approval process of medical devices in Japan
UNITED STATES
Regulatory Authority: FDA - Food and Drug Administration
The U.S. Food and Drug Administration (FDA) is the primary regulatory authority responsible for overseeing products valued at over a trillion dollars, accounting for approximately 25% of all consumer spending in the United States. Its scope includes 80% of the nation’s food supply, along with medical devices, prescription drugs, animal products, cosmetics, and tobacco products. The FDA’s Center for Devices and Radiological Health (CDRH) specifically manages the regulation of medical devices across the country. All establishments involved in medical device production and distribution are required to register with the FDA in accordance with the guidelines outlined in FDA Form 2891 and 21 CFR Part 807. With its broad regulatory reach, the FDA ensures adherence to strict standards for safety, efficacy, and quality across various sectors.
Medical device history in the United States
Regulatory advancements intended to guarantee safety and effectiveness have a significant impact on the history of medical devices in the US. The Pure Food and Drugs Act of 1906 laid the groundwork for medical device regulation by creating the Food and Drug Administration (FDA) as a consumer protection organization tasked with stopping the sale of products that were mislabeled and tampered with the 1976 Medical Device Amendments to the 1938 Food, Drug, and Cosmetic Act were established in the United States as a result of this historical background. When the new law's provisions were completely put into effect in 1978, the FDA started examining how medical devices were developed and tested in clinical settings. The 1976 Amendments have undergone numerous revisions over time, and they currently include provisions pertaining to the development of medical devices.
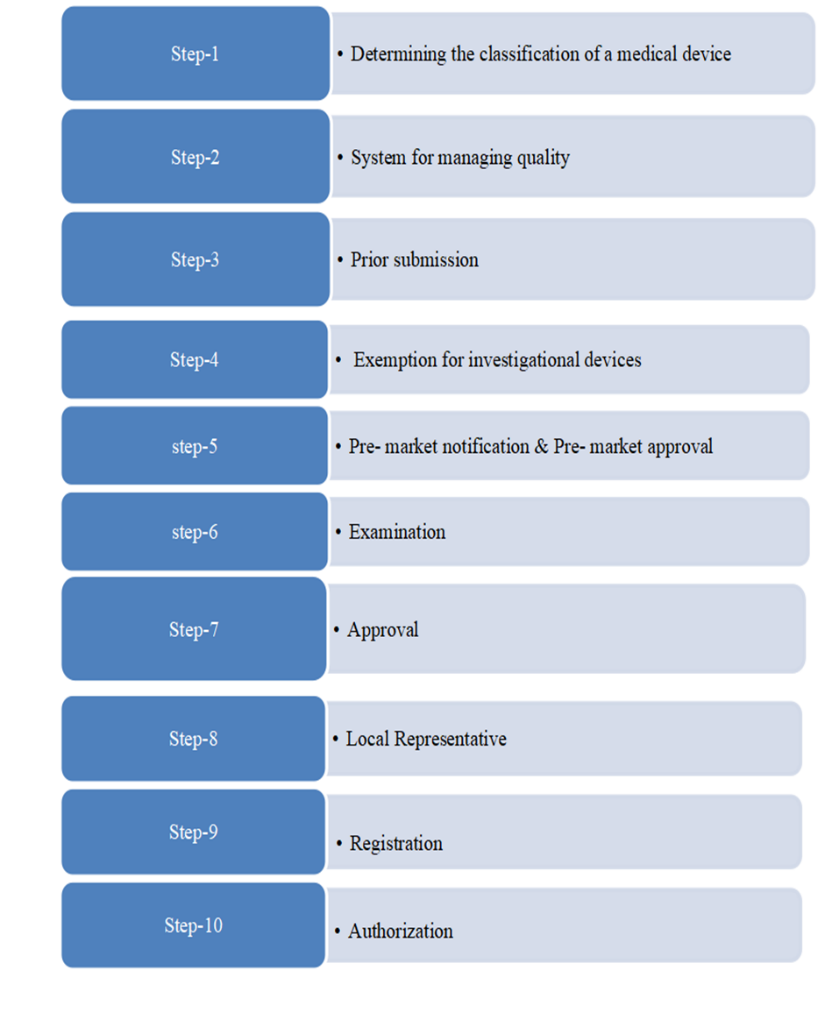
Figure:3 The process of medical devices registration in USA
The flowchart illustrates the stages involved in bringing a medical device to market and beyond. It's divided into three main sections:
Market Approval: This section focuses on the regulatory processes required to obtain market approval for the device.
Market of Device: This section covers the actual launch and commercialization of the device in the market.
Post Market Surveillanc : This section emphasizes the continuous assessment and review of the device’s safety and performance once it is available in the market.
Market Approval Section Device: This is the starting point. It represents the medical device being developed.
Class I, II, and III: These are different regulatory classes for medical devices based on their level of risk and control. Class I devices are generally low-risk, while Class III devices are high-risk and require the most stringent regulatory oversight.
510(k) Notification or PMA: These are two regulatory pathways for securing market approval. The pre market notification process is used for devices that are substantially equivalent to existing ones, whereas the PMA process is required for devices that are novel or present higher risks.
Market of Device: This signifies the introduction of the device into the market.
Post-Market Surveillance: This section covers activities like tracking adverse events, conducting post-market studies, and updating device labeling and instructions for use.
USA Medical Device Regulation
The Food and Drug Administration (FDA), in particular its Center for Devices and Radiological Health (CDRH), is principally responsible for the regulation of medical devices in the United States. The FDA's regulatory system, which was established in 1976, is intended to guarantee that medical devices are high-quality, safe, and effective during their entire existence. Class I (low risk), Class II (moderate risk), and Class III (high risk) are the three risk-level-based classifications for medical devices. While Class III devices must go through a rigorous premarket approval (PMA) process that includes proving safety and efficacy through clinical data, Class I devices usually require the least amount of regulatory scrutiny, frequently merely requiring registration with the FDA.
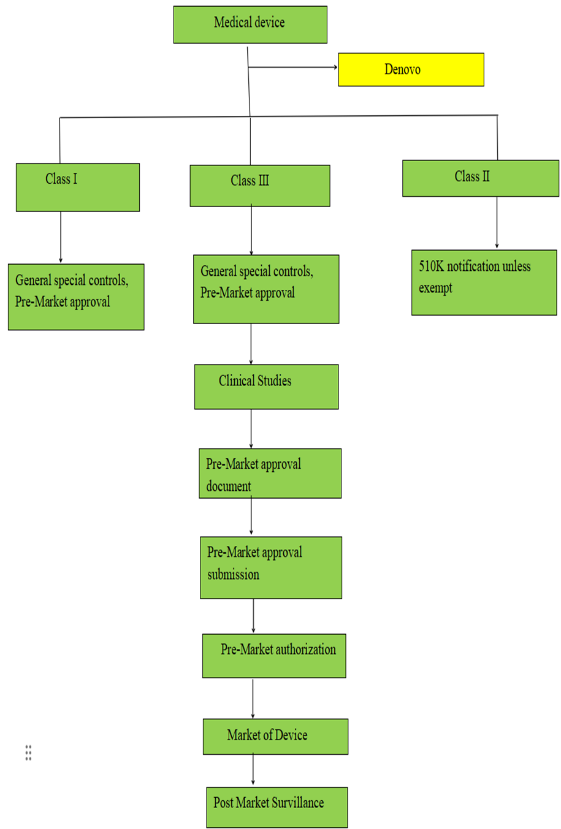
Figure:4 Approval process of medical device in USA
FDA in the US Regulation of medical devices
The U.S. Food and Drug Administration (FDA) oversees medical devices through several major roles
1. Classification: Three categories are used to classify devices.
Class I: low risk
Class II (risky but modest)
Class III (high risk)
Table:4 Medical devices classification according to USA
|
Class |
Risk level |
Examples |
|
Class- I |
Low - risk |
Wound dressings and manual surgical tools |
|
Class- II |
Moderate - risk |
Diagnostic imaging devices |
|
Class- III |
High - risk |
Pacemakers, prosthetic heart valves |
2. Premarket Review: Premarket filings, including as 510(k) notices and Premarket Approval (PMA) applications, are examined by the FDA.
3. Post-Market Surveillance: To guarantee continued safety and effectiveness, the FDA keeps an eye on a device's performance after it is put on the market through studies, recalls, and adverse event reporting.
4.Guidance and Standards: To encourage innovation and best practices, the FDA sets standards and guidelines that manufacturers must adhere to while developing, testing, and marketing medical devices.
5. Compliance and Enforcement: To guarantee compliance, the agency carries out inspections and enforces rules, taking legal action against producers who don't adhere to safety and quality requirements.
6. User Fees: To finance the regulatory process and increase efficiency, the FDA charges manufacturers fees for a range of services.
Table:5 Fee Structure by Device Class in USA
|
Applicant type |
Standard Fee for Fiscal Year 2024 |
Small Business Fee for Fiscal Year 2024 |
standard feeFY 2025 |
FY 2025 business fee |
|
510K |
$21,760 |
$5,440 |
$24,335 |
$6,085 |
|
PMA |
$483,560 |
$120,890 |
$540,783 |
$135,196 |
|
Request for DeNovo Classification |
$145,068 |
$36,267 |
$162,235 |
$40,559 |
|
180-Day Supplement |
$72,534 |
$18,134 |
$81,117 |
$20,279 |
|
Annual Fee |
$16,925 |
$4,231 |
$18,927 |
$4,732 |
3. METHODOLOGY
The United States and Japan have quite different registration and approval procedures for medical devices, and each nation has its own set of rules and procedures. The Pharmaceuticals and Medical Devices Agency (PMDA) is where applications are submitted to start the registration process in Japan. The device's intended application, design specifications, manufacturing procedures, safety and efficacy data, and details about the quality management system must all be covered in the extensive documentation needed for this initial submission. The PMDA then does a two-phase examination, first evaluating the application's accuracy and completeness and then thoroughly reviewing the device's technical and clinical aspects. The Ministry of Health, Labour and Welfare (MHLW) assesses the device's adherence to Japanese regulatory standards concurrently with the PMDA examination. Reviews of clinical data, production procedures, and quality control systems are all included in this assessment. In order to confirm compliance with Japanese rules and quality control requirements, the procedure also involves compliance and Good Manufacturing Practice (GMP) reviews. After taking into account every evaluation component, the Minister of Health, Labour, and Welfare makes the ultimate approval decision. The FDA's Center for Devices and Radiological Health (CDRH) is in charge of the regulatory procedure in the US. The process differs according on the approval procedure and device classification. Manufacturers are required to show significant equivalency to current devices in order to submit 510(k) applications. For Class III products, the Premarket Approval (PMA) procedure necessitates more thorough safety and efficacy evidence. For new low-to-moderate-risk devices without predicates, the De Novo pathway offers a path. The intricacy of these procedures is reflected in the cost schedule. Class I devices are free in Japan, while Class IV devices cost ¥147,000. The pricing structure in the U.S. system is more diverse; normal fees for FY 2024 range from $21,760 for 510(k) submissions to $483,560 for PMA applications, with small enterprises eligible for lower rates. Both nations have strong post-market surveillance programs in place to keep an eye on the performance and safety of approved devices, guaranteeing continued adherence to legal requirements and safeguarding the public's health.
Flow Process of study
Table: 6 COMPARISION OF REGULATORY ASPECTS JAPAN AND USA
|
Sr. No. |
Aspect |
JAPAN |
USA |
|
1. |
Regulatory Authority |
PMDA |
FDA |
|
2. |
Classification |
Class I,II,III,IV |
Class I,II,III |
|
3. |
Approval Pathway |
PMDA, MHLW |
510(k), PMA, DeNovo |
|
4. |
Clinical Trials |
Required for most Class II and III devices, including local trials |
Required for Class III, some Class II devices |
|
5. |
Timeline |
6 Months- several years |
510(k) submissions take about 3-6 months; PMA can take several years |
|
6. |
Post-Market Surveillance |
Mandatory post-market surveillance for all devices, including adverse event reporting |
Well-established system with MDR, GUDID |
|
7. |
Local Agent Requirement |
Required; manufacturers must appoint a local Marketing Authorization Holder (MAH) |
Not required; manufacturers can register directly with the FDA. |
|
8. |
Fees Structure |
Fees vary based on the type of submission |
Fees vary based on the type of submission (510(k) or PMA) |
DISCUSSION
The analysis of medical device approvals and rejections in the USA and Japan reveals distinct patterns in their regulatory approaches:
In the USA:
Table:7 Approved applications in USA
|
Class |
2022 |
2023 |
2024 |
|
510K |
1,726 |
1,817 |
1,776 |
|
PMA |
19 |
35 |
40 |
|
DENOVO |
79 |
68 |
30 |
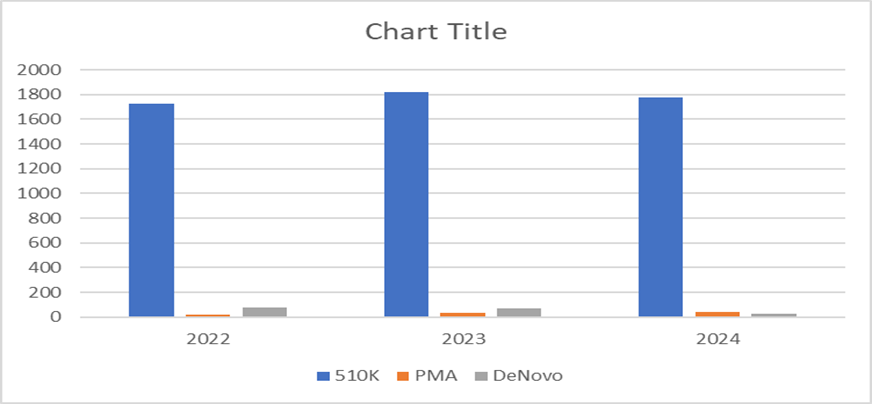
Graph: 5 Bar graph of approved appliacations in USA
TABLE:8 Rejected applications in USA
|
Class |
2022 |
2023 |
2024 |
|
510K |
428 |
454 |
444 |
|
PMA |
8 |
23 |
20 |
|
DENOVO |
10 |
12 |
3 |
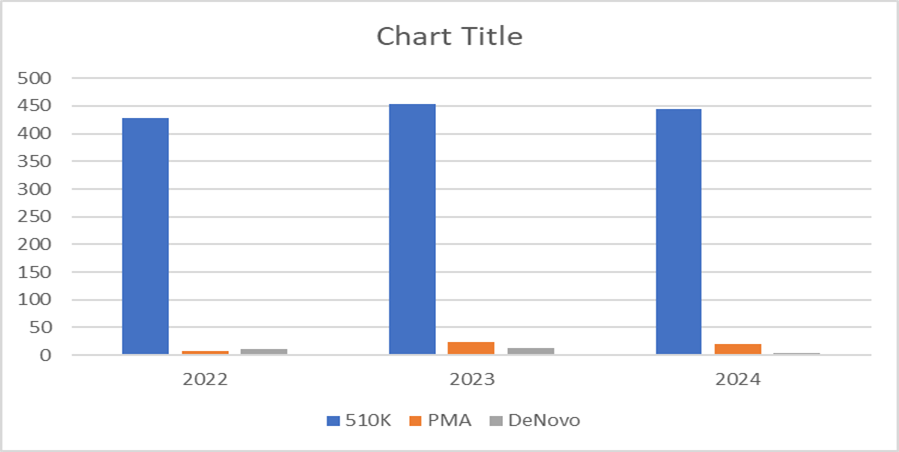
Graph: 6 Bar graph of rejected applications in USA
In Japan:
Table:9 Approved applications in Japan
|
Class |
2022 |
2023 |
2024 |
|
Class I |
1,198 |
1,302 |
1,359 |
|
Class II |
2,076 |
2,223 |
2,334 |
|
Class III |
1,345 |
1,456 |
1,542 |
|
Class IV |
634 |
678 |
724 |
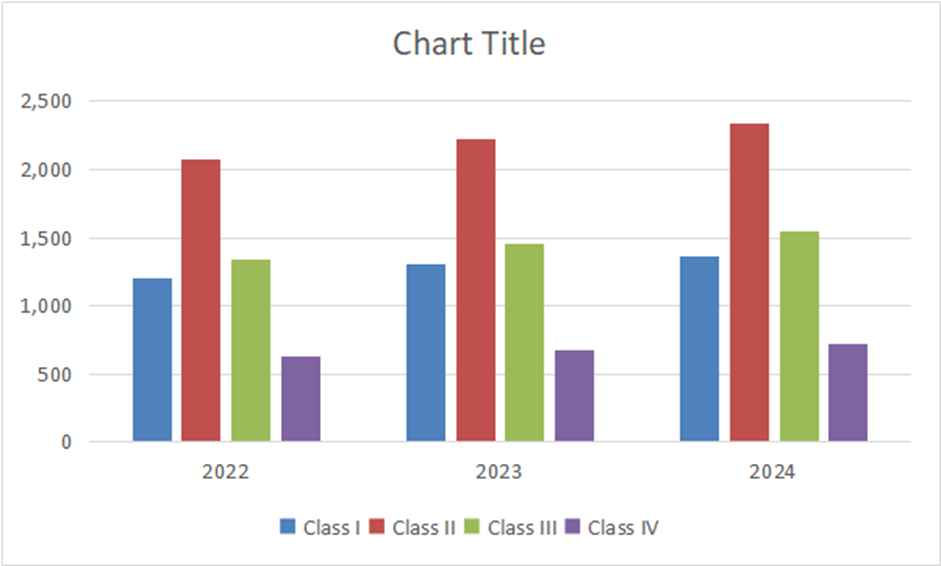
Graph:7 Bar graph of apporved applications in Japan
Table:10 Rejected applications in Japan
|
Class |
2022 |
2023 |
2024 |
|
Class I |
47 |
54 |
53 |
|
Class II |
111 |
122 |
122 |
|
Class III |
111 |
131 |
147 |
|
Class IV |
122 |
134 |
152 |
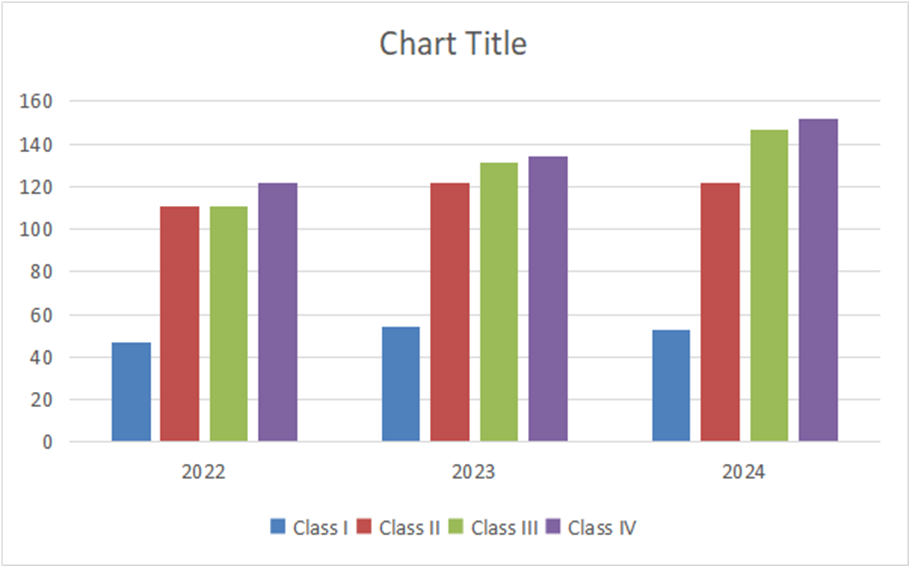
Graph: 8 Bar graph of rejected applications in Japan
The comparison highlights that while the USA's 510(k) process facilitates faster market entry for moderate-risk devices, Japan's system emphasizes more thorough evaluations across all risk categories. Both countries show increasing approval trends, suggesting growing medical device remaining relatively stable across the years.Innovation while maintaining their respective safety standards.
SUMMARY
This document provides a comparative review of the registration and approval processes for medical devices in the United States and Japan. Both nations, being major players in the global healthcare industry, have established robust regulatory frameworks to ensure the safety, efficacy, and quality of medical devices while encouraging innovation. The U.S. Food and Drug Administration (FDA) employs a risk-based three-tier classification system, including pathways like 510(k), Premarket Approval (PMA), and De Novo processes. These pathways are designed to facilitate market entry based on the device's risk level. The Japanese Pharmaceuticals and Medical Devices Agency (PMDA), operating under the Ministry of Health, Labour, and Welfare (MHLW), uses a four-tier classification system and emphasizes clinical trials for higher-risk devices. While the U.S. process promotes efficiency through its streamlined 510(k) approvals, Japan's system ensures thorough evaluations with stringent reviews for high-risk devices. Statistical trends from 2022 to 2024 reveal that the U.S. achieves high annual approval volumes, particularly through the 510(k) pathway, reflecting substantial equivalence to existing devices. In contrast, Japan demonstrates steady growth in Class II device approvals, showing its focus on rigorous oversight. Both nations face challenges like high costs, complex regulations, and the need for global harmonization, but their approaches highlight different priorities: the U.S. leans towards speed, while Japan emphasizes safety. Future directions emphasize enhancing global collaboration, harmonizing regulatory frameworks, and improving post-market surveillance to balance innovation and public health. These insights are vital for stakeholders navigating these significant healthcare markets.
CONCLUSION
The comparative analysis of medical device regulatory frameworks in the USA and Japan highlights key similarities and differences in their approval processes, classification systems, and post-market surveillance mechanisms. The U.S. FDA employs a three-tier classification system with streamlined 510(k) pathways that facilitate faster market entry for moderate-risk devices, processing over 1,700 approvals annually. In contrast, Japan's PMDA uses a four-tier classification system.In USA 510k has higher approval rate in the year 2022-2024 compared to PMA and Denovo applications. In Japan Class II devices have over 2,000 approvals annually and steady growth across all device classes. In USA 510K applications have higher rejection rate compared to PMA nad Denovo applications, whereas japan has higher rejection rate for class III and IV compared to class I,II. This data says how both systems effectively balance innovation with public safety through their distinct regulatory frameworks. Both the United States and Japan face challenges in their medical device regulatory frameworks, including high approval costs, regulatory complexity, and the need for harmonization to overcome these issues requires that improving post-market surveillance systems and leveraging advanced technologies could enhance monitoring and ensure continued device efficacy and investing in advanced technologies like AI for regulatory review and expanding training programs for manufacturers on compliance can improve efficiency.
ACKNOWLEDGEMENTS
I would like to thank Mr. Sandeep Kanna for his advice and assistance in seeing this article through to completion, as well as the Chalapathi Institute of Pharmaceutical Sciences for their steadfast support.
FUNDING: None to declare
CONFLICT OF INTEREST: None to declare
REFERENCES


Kunda Vandana, Dr. Sandeep Kanna, Comparison of Medical Device Regulatory Frameworks: Approval and Registration Processes in the Usa and Japan, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 2279-2290. https://doi.org/10.5281/zenodo.15975390
 10.5281/zenodo.15975390
10.5281/zenodo.15975390