
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Adhiparasakthi College of Pharmacy, Melmaruvathur, Tamil Nadu 603319.
Chalcones, a class of naturally occurring and synthetically derivable flavonoids, have shown promising pharmacological properties, including anxiolytic activity. In the present study, a series of benzimidazole-based chalcone analogues were evaluated as potential anxiolytic agents using an integrated in silico and in vivo approach. Ten chalcone derivatives were designed, synthesized, and assessed for their binding affinity to two key target proteins (PDB IDs: 6CDU and 6IQL) using molecular docking tools (AutoDock Vina in PyRx and Argus Lab). Diazepam served as the reference drug. Drug-likeness and ADMET properties were predicted using Molinspiration and SwissADME to assess oral bioavailability and pharmacokinetic compatibility. Docking studies revealed that compound CC exhibited the highest binding affinity with 6CDU (-13.92 kcal/mol) and 6IQL (-12.24 kcal/mol), surpassing the reference drug. All compounds complied with Lipinski’s rule of five, showed high gastrointestinal absorption, and demonstrated favorable BBB permeability. The findings suggest that benzimidazole-based chalcones, particularly compound CC, possess significant potential as anxiolytic agents with favorable pharmacokinetic profiles.
“Chalcones” were discovered by Kostanecki and Tambor. They are supernumerary present in nature from ferns to higher plants and polyhydroxylated in the aryl rings present in the plants. Chalcones are changed into flavonoids catalyzed by the enzyme chalcone isomerase. These are naturally present in variety of plant species like Angelica, Glycyrrhiza Humulus and Scutellaria. The substances are widely present in the plant (fruits, vegetables, soy-based food stuff). These are supernumerary in comestible plants. The most common chalcones present in foods are phloretin and its glucosidephloridzin (phloretin 2’- o-beta –glucopyranoside) and chalconaringenin. Chalcones are among the leading categories of flavonoids across the entire kingdom of plant. They are also recognized as benzylidene acetopheonone. Chalcone is an alpha, beta unsaturated ketone consisting of two benzenoid rings (ring A and ring B)with wide variety of groups. Aromatic groups are connected to each other by three carbons, α,β- unsaturated ketonic system, highly electrophilic in nature having a linear structure. They have ketoethylenic moiety(–CO–CH=CH–) and a conjugated double bond. The chemical structure of chalcones contains several replaceable hydrogens that allow a large number of derivatives to be obtained. Chalcones incorporating heterocyclic scaffolds have been reported with various biological and pharmacological activities such as antidepression, antimicrobial, antimalarial, antifungal, anthelmentic, anxiolytic, analgesic, anxiolytic antinociceptive, anti-HIV, monoamino oxidase inhibition, antiangiogenic, anti-leshmanial activities etc.[3,7]
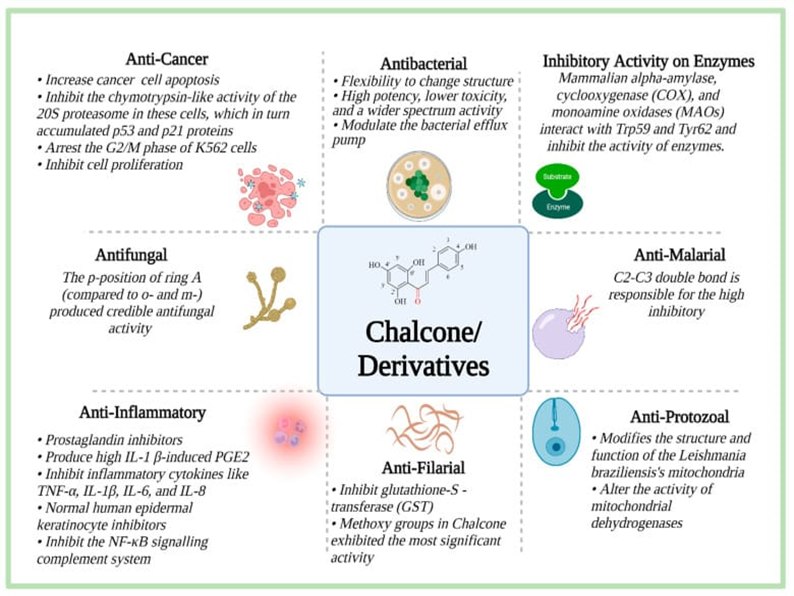
Figure 1 Pharmacological properties of Chalcones
Chemically chalcones are easily prepared using various reaction procedures and strategies. Claisen-Schmidt condensation is one common methodology to prepare the chalcone through carbonyl derivative condensation in the presence of base. Additionally, the carbonalylative Heck coupling reaction, the Sonogashira isomerization coupling reaction, the continous flow deuteration reaction, the Suzuki-Miayura coupling reaction, and solid acid catalyst-mediated reactions are known. A series of benzimidazole based chalcone derivatives were synthesized and reported using the Claisen-Schimdt reaction.[6]
ANXIETY:
Psychotic behaviors are characterized by disturbances of reality and perception, impaired cognitive functioning and mood disturbances. There are four major psychotic disorders that are commonly encountered in clinical practice. They are anxiety disorders, mood disorders, personality disorders and somatoform disorders.
ANXIETY DISORDERS[13]
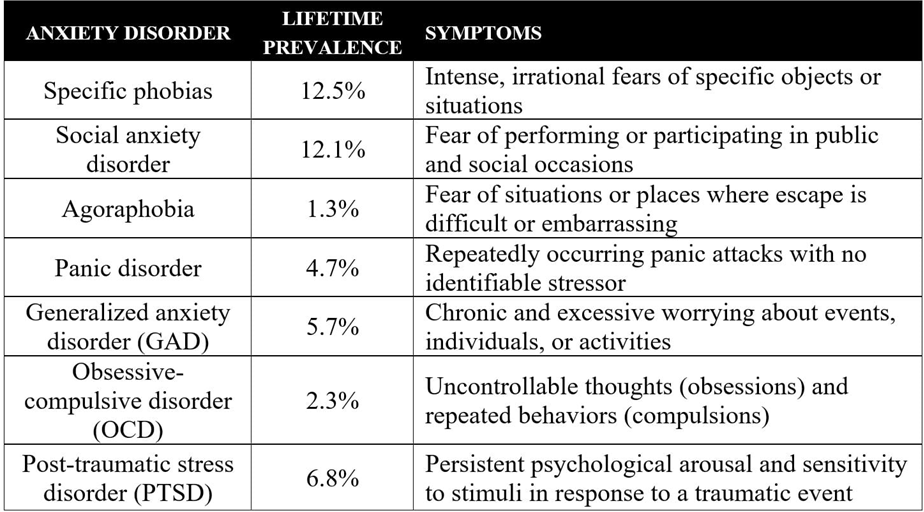
Anxiety is defined as a subjective sense of unease, dread (fear with terror) or foreboding. It may be a primary psychiatric disorder or due to some organic disease. There are five types of anxiety disorders.
Panic disorders: Panic disorders are recurrent and unpredictable attacks of fear and discomfort associated with a variety of physical symptoms such as palpitations, pounding heart, sweating, trembling, sensations of shortness of breath, choking sensation, chest pain and discomfort, nausea or abdominal distress, dizziness, unsteadiness, feeling of unreality, fear of dying, parathesia, chills and hot flashes, and agarophobia (situational anxiety where escape is difficult).
Generalized anxiety disorders: generalized anxiety disorders comprise excessive anxiety even for carrying out smaller tasks, unexplained worry, restlessness, fatigue, difficulty in concentrating, mental stress, irritability and sleep disturbances. It is neither due to a direct psychiatric effect nor due to any organic disease.
Phobic disorders: They are manifested as marked and persistent fear o objects or situations, exposure too which results in immediate anxiety reaction. The affected person avoids the phobic stimulus and this avoidance usually impairs occupational or social functions. Here the patients experience anxiety only in specific and identical situations-fear of closed spaces(claustrophobia), fear of blood, fear of flying and social phobia.
Stress disorders: In such cases patients develop significant anxiety due to exposure to extreme situations like trauma, accident and disease.
Obsessive-compulsive disorder: Here the affected persons experience obsessive thoughts and compulsive behavior such as fear of contamination and infection, checking and rechecking it regard to locking doors, counting currency notes or washing of hands repeatedly.
Table1 Lifetime Prevalence of Different Anxiety Disorders
Anxiety is an emotional state, unpleasant in nature, associated with uneasiness, discomfort and concern or fear about some defined or undefined future threat. Some degree of anxiety is a part of normal life. Treatment is needed when it is disproportionate to the situation and excessive. Some psychotics and depressed patients also exhibit pathological anxiety. Anxiolytics are medications that can treat anxiety and related conditions. The bestknown anxiolytics are benzodiazepines and barbiturates. They have many associated toxic effects, such as sedation, drowsiness, vertigo, mood swings, paronia, irritability, and neurotoxicity.[2] Anxiety and depression are disorders that cause substantial economic burden and require new treatments with better therapeutic efficacy, fewer adverse effects and a short latency. The anxiolytic effect shown by chalcones increased strength, low cost, superior pharmacokinetic properties, and minimum side effects.
MECHANISM OF ACTION OF ANXIOLYTICS[5]

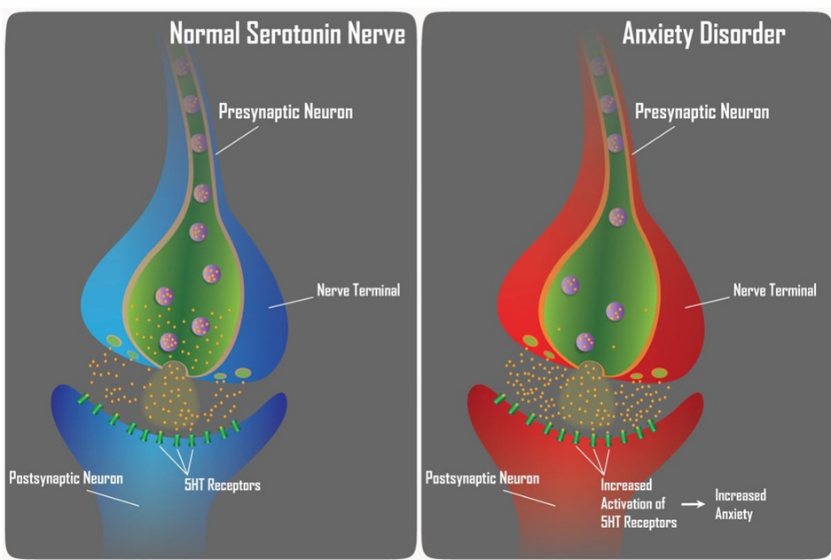
Figure 2 Hypothesis of ANXIETY disorders
DRUG DISCOVERY
The process of finding novel drugs to treat different illnesses is called drug discovery. Target identification, lead discovery, lead optimization, preclinical testing, clinical trials, and regulatory approval are some of the phases in this lengthy and complex process.
STEPS OF DRUG DISCOVERY AND DEVELOPMENT
1. TARGET IDENTIFICATION
Target identification is the initial phase of drug discovery. A biological target is as an element or technique that has an effect in a certain illness. Any biological mechanism essential to development of the disease, whether it be a protein, enzyme, gene, or any other, might be the cause of the disease. Potential targets are discovered by scientists using a variety of methods, including genetic and biochemical methodologies, epidemiological research, and clinical observations.
2. LEAD DISCOVERY
The subsequent stage in drug development is lead discovery, which comes after a potential target has been located. A chemical that interacts with the targets and has positive pharmacological action is called a lead compound.
3. LEAD OPTIMIZATION
Lead optimization is the subsequent phase of drug development after the identification of lead molecules. To increase the lead compound’s pharmacological activity, selectivity, and safety, the structure must be modified. Lead optimization can be done using a variety of methods, including as medicinal chemistry, structural biology, and pharmacology.
DOCKING STUDIES
Docking is a method which predicts the preferred orientation of one molecule to a second when bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules. Molecular docking is one of the most frequently used methods in structure-based drug design, due to its ability the binding-conformation of small molecule ligands to the appropriate target binding site.
Various types of docking in the basis of the possible different molecular interactions,
MOLECULAR DOCKING[12]
Molecular docking is a key tool in structural molecular biology and computer assisted drug design. The goal of ligand –protein docking is to predict the predominant binding mode(s) of a ligand with a protein of known three-dimensional structure. Molecular docking may be defined as an optimization proble, which would describe the ‘bestfit’ orientation of a ligand that binds to a particular protein of interest. However, since both the ligand and the protein are flexible, a “hand in glove” analogy is a more appropriate than “lock-and-key”. During the course of the process, the ligand and the protein adjust the conformation to achieve an overall “best fit” and this kind of conformational adjustment resulting in overall binding is referred to as an “induced-fit”. On molecular docking using software are GLIDE, M AUTODOCK, GOLD, SULFLEX, FLEX, LIGANDFIT, GLANDDOCK, PYRX, ARGUSLAB.
STEPS IN MOLECULAR DOCKING
KEY STAGES IN DOCKING
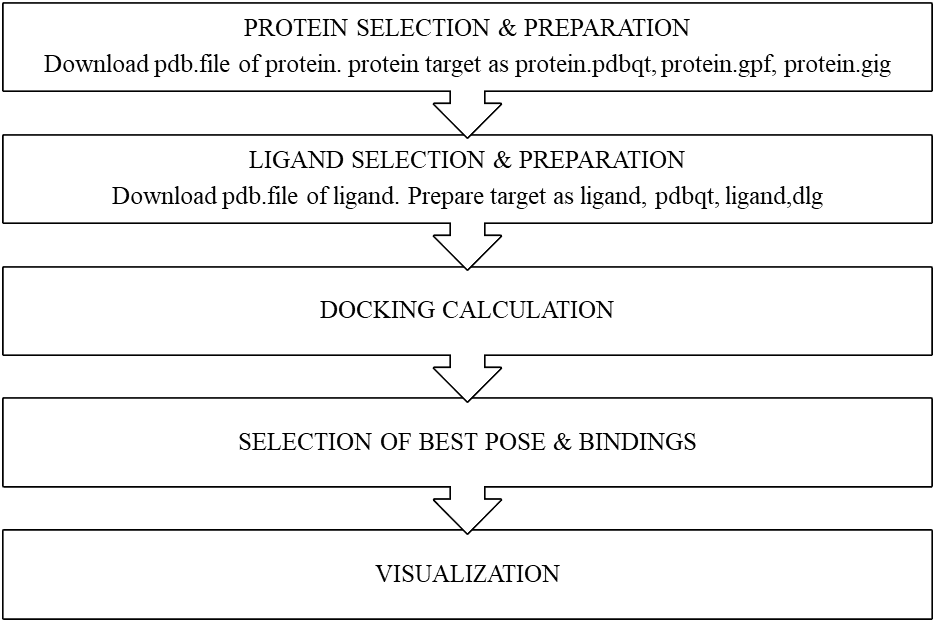
PROCESS OF MOLECULAR DOCKING:
The 3-D structure for the target macromolecules and the smaller molecules must be first choosen and then each structure must be prepared in accordance with the requirements of the docking method being used.
The problems that arise with the use of antianxiety substances are adverse effects. To overcome the adverse effects, several chemicals will be synthesized and tested for anxiolytic activity. Since synthesizing and testing of antianxiety activity by neuropharmacological evaluation protocols is a tedious and time-consuming process here docking software is used for the prediction of anxiolytic activity. The computer software used in the present studies is
ACD/ CHEMSKETCH:
ACD/ Chemsketch is a powerful all-purpose chemical drawing and graphics package from ACDlabs to help chemist quickly and easy to draw molecular structures, reactions, schematic diagrams, calculate chemical properties and design professional report and presentations. It includes:
ARGUS LAB: Argus lab is a docking software. Docking is computational stimulation of a candidate ligand binding to a receptor. In binding mode, the orientation of the ligand relative to the receptor as well as the conformation of the ligand and receptor is adjusted when bound to each other. Scoring is the process of evaluating a particular pose by counting the number of favorable intermolecular reactions such as hydrogen bonds and hydrophobic contacts.
DISCOVERY STUDIO:
Discovery studio is a suite of software for simulating small molecule and macromolecule systems. It is developed and distributed by Dassault Systems BIOVIA.
Discovery studio provides software applications covering the following areas: Simulations, Ligand design, Pharmacophore modeling, Structure-based design, Macromolecule design and validation, Macromolecule engineering, QSAR, ADME, Predictive toxicity.
BIOLOGICAL SCORE:
Bioactivity of drugs can be checked by calculating the activity score of ligands, and GABA receptor.
All the parameters will be checked with the help of software molinspiration drug likeness score online (www.molinspiration.com). Calculated drug likeness score of each compound and compared the specific activity of each compound, and the results were compared with standard drug.
For organic molecules the probability of the bioactivity score is (>0). Then it is active if (-5.0 to 0) then moderately active, if (< -5.0) then inactive. Molinspiration is based on Lipinski’s rule.
LIPINSKI’S RULE:
Lipinski’s rule of five also known as the Pfizer’s rule of five or simply the rule of five (RO5) is a rule of thumb to evaluate drug likeness are determined if a chemical compound with certain pharmacological or biological activity as properties that would make a likely orally active drug in humans.
The rule was formulated by Christopher A. Lipinski in 1977. The rule describes molecular properties important for a drug’s pharmacokinetics in human body, including the Absorption, Distribution, Metabolism, and Excretion (“ADME”) components of the Lipinski’s rule.
LIPINSKI’S RULE STATE
PREDICTION OF ADMET BY SWISS ADME
Pharmacokinetic properties such as absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiling of compounds were determined using the ADMET descriptors algorithm protocol, the lipophilicity levels in the form of atom-based LogP. The absorption of drugs depends on factors including membrane permeability, intestinal absorption, skin permeability levels, P-glycoprotein substrate, or inhibitor. The distribution of drugs depends on factors that include the blood brain barrier, CNS permeability, and the volume of distribution. Metabolism is predicted based on the CYP models for substrate or inhibition (CYP2D6, CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). Excretion is predicted based on the total clearance model and renal OCT2 substrate. The toxicity of drugs is predicted based on AMES toxicity, hERG inhibition, hepatoxicity, and skin sensitization. These parameters were calculated and checked for compliance with their standard ranges.
MATERIALS AND METHODS
Preparation of protein
Three-dimensional structure of the protein should be retrieved from the RCSB Protein data bank(PDB); afterward the retrieved structure should be pre-processed for removal of heteroatoms then energy minimization was performed by using Argus lab. Then the energy-minimized structure was saved for further use.
Preparation of ligand
Ligands can be retrieved from several databases such as ZINC, PubChem or can be sketched by applying the Chemsketch tool. Ligand 2D structures were drawn using ACD Chemsketch. The crystal structures of two target proteins (PDB Codes: 6IQL, 6CDU) were retrieved from the RCSB Protein Data Bank in PDB file format, and then prepared separately by eliminating water molecules, cofactors and co-crystallised ligands contained within the protein structures. The drug molecule Diazepam was collected in 3D SDF format from the PubChem database.
Table 1 Designed benzimidazole chalcones using ACD Chemsketch
|
Sr.No |
Compound |
Structure |
IUPAC NAME |
|
1. |
AC |
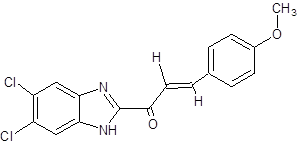
|
(2E)-1-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)-3-(4-methoxyphenyl)prop-2-en-1-one |
|
2. |
BC |
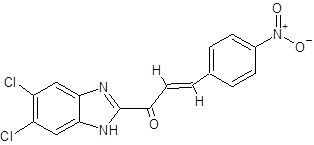
|
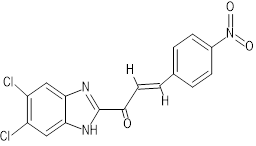
(2E)-1-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)-3-(4-nitrophenyl)prop-2-en-1-one |
|
3. |
CC |
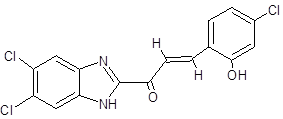
|
(2E)-3-(4-chloro-2-hydroxyphenyl)-1-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)prop-2-en-1-one |
|
4. |
DC |
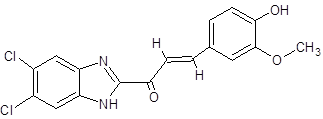
|
(2E)-1-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)-3-(4-hydroxy-3-methoxyphenyl)prop-2-en-1-one |
|
5. |
EC |
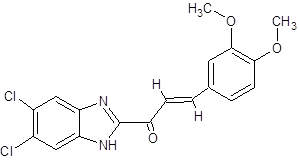
|
(2E)-1-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one |
|
6. |
FC |
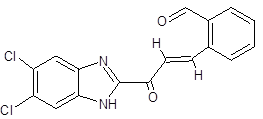
|
2-[(1E)-3-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)-3-oxoprop-1-en-1-yl]benzaldehyde |
|
7. |
GC |
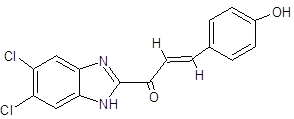
|
(2E)-1-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)-3-(4-hydroxyphenyl)prop-2-en-1-one |
|
8. |
HC |
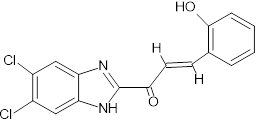
|
(2E)-1-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)-3-(2-hydroxyphenyl)prop-2-en-1-one |
|
9. |
IC |
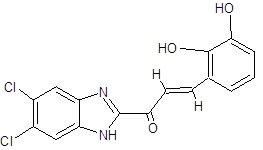
|
(2E)-1-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)-3-(2,3-dihydroxyphenyl)prop-2-en-1-one |
|
10. |
JC |
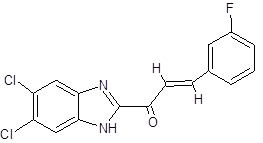
|
(2E)-1-(5,6-dichloro-1H-1,3-benzimidazol-2-yl)-3-(3-fluorophenyl)prop-2-en-1-one |
Docking
Molecular docking investigation was performed separately between the two proteins and all 10 compounds, including the reference drug (Diazepam) using the AutoDock Vina of PyRx software tool and Argus lab. The screening was conducted to ascertain the most active benzimidazolechalcone compounds against the protein targets. PyRx calculates the binding affities of the receptor-ligand interactions which are necessary to describe how fit the molecules bind to the target protein. A more negative binding affinity will indicate a greater chance of the potential drug molecule to initiate protein biochemical action/reaction.
Evaluation of pharmacokinetic properties
Predicting pharmacokinetics properties plays a critical role in the early stage of drug discovery. This is because only molecules which demonstrate good ADMET and drug-likeness properties reach the preclinical research phase. The compounds were subjected to drug-likeness and ADMET tests using online web server http://www.swissadme.ch/index.php. Lipinski’s rule of five (RO5) also called the Pfizer rule is a well-established provision for determining the oral bioavailability of a given compound. Consequently, these analogs were subjected to the RO5 criterion to ascertain their oral bioavailability.
RESULTS AND DISCUSSION
Virtual Docking Screening
Molecular docking simulations were conducted to assess the binding affinity of ten benzimidazole-based chalcone derivatives against two target proteins associated with anxiolytic activity (PDB IDs: 6CDU and 6IQL). Diazepam served as the reference drug. The docking scores obtained from AutoDock Vina (via PyRx) and Argus Lab are presented in Table 1.
Docking results revealed that compound CC exhibited the strongest interaction with both protein targets, achieving a binding affinity of -13.92 kcal/mol for 6CDU and -12.24 kcal/mol for 6IQL. These values were significantly more negative than those recorded for Diazepam, indicating a stronger predicted binding. Other derivatives, notably GC (-12.23 kcal/mol with 6IQL) and FC (-12.15 kcal/mol with 6IQL), also demonstrated high binding affinities.
In general, most chalcone derivatives displayed better docking scores than Diazepam, suggesting that their structural features—such as the benzimidazole moiety coupled with the chalcone scaffold—enhanced receptor binding. The higher negative binding energies suggest that these molecules fit snugly within the active sites of the target proteins, potentially leading to higher pharmacological efficacy.
Table1 Summary of binding affinities interactions between benzimidazolechalocne derivatives and different protein targets using argus lab
|
Sr. No. |
Compound |
Binding Energy With 6cdu (Kcal/Mol) |
Binding Energy With 6iql (Kcal/Mol) |
|
1. |
AC |
-10.8674 |
-11.0931 |
|
2. |
BC |
-6.4302 |
-10.6774 |
|
3. |
CC |
-13.9217 |
-12.2429 |
|
4. |
DC |
-10.4614 |
-10.5368 |
|
5. |
EC |
-8.55683 |
-11.0759 |
|
6. |
FC |
-9.35496 |
-12.1505 |
|
7. |
GC |
-8.06756 |
-12.2355 |
|
8. |
HC |
-6.1496 |
-11.5771 |
|
9. |
IC |
-5.88867 |
-10.1563 |
|
10. |
JC |
-10.1097 |
-11.499 |
Table 3 Summary of binding affinities interactions between some benzimidazolechalocne derivatives and protein target 6CDU using Pyrx
|
Sr. No. |
Strucure |
Docking Image |
Binding Score |
|
Std 1 |
|
|
-7.7 |
|
|

|
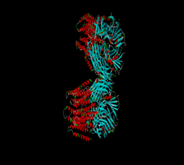
|
-8.9 |
|
|
|

|
-9.2 |
|
|
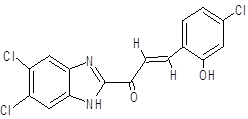
|

|
-9.2 |
Table 4 Summary of binding affinities interactions between some benzimidazolechalocne derivatives and protein target 6IQL using Pyrx
|
SR. NO. |
STRUCTURE |
DOCKING IMAGE |
SCORE |
|
Std 1 |
|

|
-8.9 |
|
|
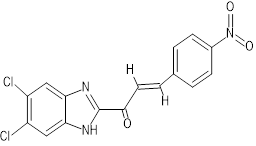
|

|
-11 |
|
2. |
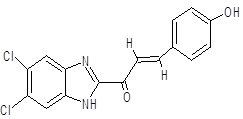
|
|
-10.1 |
|
3. |
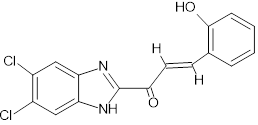
|
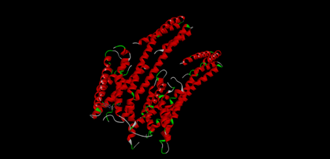
|
-10.1 |
Table 4 Predicted drug-likeness properties of benimidazolechalcone derivatives
|
Sr. No. |
Compound |
Molecular weight |
No. of heavy atoms |
No. of aromatic heavy atoms |
Fraction csp3 |
No. Of rotatable bonds |
No. Of h-bond acceptors |
No.of h bond donors |
Molar refractivity |
Tpsa (A2) |
Logp o/w |
Log s |
|
1. |
AC |
349.21 |
23 |
12 |
0.12 |
4 |
2 |
2 |
99.94 |
50.36 |
2.97 |
-5.19 |
|
2. |
BC |
364.18 |
24 |
12 |
0.06 |
4 |
3 |
2 |
102.27 |
89.95 |
1.71 |
-5.18 |
|
3. |
CC |
369.63 |
23 |
12 |
0.06 |
3 |
2 |
3 |
100.48 |
61.36 |
2.49 |
-5.57 |
|
4. |
DC |
365.21 |
24 |
12 |
0.12 |
4 |
3 |
3 |
101.96 |
70.59 |
2.79 |
-5.05 |
|
5. |
EC |
379.24 |
25 |
12 |
0.17 |
5 |
3 |
2 |
106.43 |
59.59 |
2.68 |
-5.26 |
|
6. |
FC |
347.20 |
23 |
12 |
0.06 |
4 |
2 |
2 |
98.84 |
58.20 |
2.44 |
-4.86 |
|
7. |
GC |
335.18 |
22 |
12 |
0.06 |
3 |
2 |
3 |
95.47 |
61.36 |
2.21 |
-4.98 |
|
8. |
HC |
335.18 |
22 |
12 |
0.06 |
3 |
2 |
3 |
95.47 |
61.36 |
2 |
-4.98 |
|
9. |
IC |
351.18 |
23 |
12 |
0.06 |
3 |
3 |
4 |
97.49 |
81.59 |
1.77 |
-4.84 |
|
10 |
JC |
337.18 |
22 |
12 |
0.06 |
3 |
2 |
2 |
93.41 |
41.13 |
2.60 |
-5.28 |
Drug-Likeness Evaluation
Drug-likeness properties were determined according to Lipinski’s Rule of Five (RO5) to predict oral bioavailability (Table 5). All compounds complied with RO5, showing zero violations. Molecular weights were below 500 Da, hydrogen bond donors ≤ 5, and hydrogen bond acceptors ≤ 10.
The Topological Polar Surface Area (TPSA) values were all below 100 Ų, indicating good permeability across biological membranes, including potential blood-brain barrier (BBB) penetration. The calculated Log P values (1.71–2.97) indicated moderate lipophilicity, a desirable property for central nervous system (CNS)-active agents, ensuring both solubility and membrane penetration.
Table 5 Predicted ADMET properties benzimidazolechalcone derivatives
|
Sr. No. |
Compound |
GI absorption |
BBB permeation |
Pgp substrate |
CYP1 A2 |
CYP2 C19 |
CYP2 C9 |
CYP2 D6 |
CYP 3A |
Bioavailability score |
Lipinski’s violation |
|
1. |
AC |
HIGH |
YES |
NO |
YES |
YES |
YES |
YES |
YES |
0.55 |
0 |
|
2. |
BC |
HIGH |
NO |
NO |
YES |
YES |
YES |
NO |
NO |
0.55 |
0 |
|
3. |
CC |
HIGH |
YES |
NO |
YES |
YES |
YES |
YES |
YES |
0.55 |
0 |
|
4. |
DC |
HIGH |
YES |
NO |
YES |
YES |
YES |
YES |
YES |
0.55 |
0 |
|
5. |
EC |
HIGH |
YES |
NO |
YES |
YES |
YES |
YES |
YES |
0.55 |
0 |
|
6. |
FC |
HIGH |
YES |
NO |
YES |
YES |
YES |
YES |
YES |
0.55 |
0 |
|
7. |
GC |
HIGH |
YES |
NO |
YES |
YES |
YES |
YES |
YES |
0.55 |
0 |
|
8. |
HC |
HIGH |
YES |
NO |
YES |
YES |
YES |
YES |
YES |
0.55 |
0 |
|
9. |
IC |
HIGH |
NO |
NO |
YES |
YES |
NO |
YES |
YES |
0.55 |
0 |
|
10. |
JC |
HIGH |
YES |
NO |
YES |
YES |
YES |
YES |
YES |
0.55 |
0 |
ADMET Prediction
ADMET profiling using SwissADME indicated that all chalcone derivatives possessed high gastrointestinal (GI) absorption (Table 3). Most compounds showed positive BBB permeability, supporting their potential to exert effects on CNS targets.
Importantly, none of the derivatives were predicted to be P-glycoprotein (P-gp) substrates, reducing the likelihood of efflux from brain tissue and enhancing CNS availability. Interaction with cytochrome P450 (CYP) isoenzymes was noted but without violating drug-likeness parameters, suggesting predictable metabolic behavior. The bioavailability score was uniform across all derivatives (0.55), further indicating good oral drug potential.
CONCLUSION
The present study identified benzimidazole–chalcone derivatives as promising candidates for anxiolytic drug development through integrated in silico evaluation. Molecular docking revealed that several derivatives, particularly compound CC, demonstrated stronger predicted binding affinities to target proteins than the reference drug Diazepam. All compounds complied with Lipinski’s Rule of Five, showed favorable ADMET profiles, high gastrointestinal absorption, and potential blood–brain barrier permeability. These findings suggest that benzimidazole–chalcones, especially CC, warrant further in vivo pharmacological evaluation to confirm their efficacy and safety as novel anxiolytic agents.
ACKNOWLEDGEMENT:
The authors wish to thank Sakthi Arul Thiru Amma and Thirumathi Amma ACMEC Trust, for providing facilities to do the working successful manner. We are grateful to thank our Dean Research and Director Academic Prof. Dr. T. Vetrichelvan, M.Pharm., Ph.D., and our principal Dr. D. Nagavalli, M.Pharm., Ph.D., for the kind support and encouraging for the completion of work.
REFERENCES

Priyadharshini E, Dr. G. Abirami, Identification of Some Chalcone Analogues as Potential Anxiolytic Agents: An Integrated In-Silico Evaluation, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 2589-2602. https://doi.org/10.5281/zenodo.16933985
 10.5281/zenodo.16933985
10.5281/zenodo.16933985