
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,2,3,4,5,6 S.C.S College of Pharmacy, Harapanahalli
7 Y.S.S College of Pharmacy, Kudligi
Transferosomes are ultra-deformable, lipid-based vesicular carriers that have revolutionized transdermal drug delivery by overcoming the limitations posed by the stratum corneum barrier. Composed of phospholipids and edge activators such as surfactants or bile salts, transferosomes exhibit exceptional elasticity, enabling them to squeeze through pores much smaller than their own diameter while maintaining structural integrity. This review highlights their formulation principles, mechanisms of penetration, and physicochemical characteristics, along with various preparation and evaluation techniques. Compared to conventional vesicular systems like liposomes, niosomes, and ethosomes, transferosomes demonstrate superior skin permeation, high drug entrapment efficiency, biocompatibility, and the capability to encapsulate both hydrophilic and lipophilic drugs. They have shown significant potential in delivering peptides, proteins, corticosteroids, insulin, anesthetics, NSAIDs, anticancer, and herbal drugs, enhancing therapeutic efficacy and patient compliance. Despite limitations such as stability issues and high production costs, transferosomes offer a promising approach for controlled and targeted transdermal drug delivery. Continuous research and optimization could further advance their role in non-invasive, effective, and patient-friendly therapeutic systems.
The skin acts as a natural barrier, protecting our bodies from the outside world. Interestingly, this same barrier can also be used to deliver medications through transdermal methods. This approach helps avoid the side effects often associated with oral drugs, can improve how well a drug works, and often makes treatment easier for patients to stick with. However, not all drugs are suitable for delivery through the skin. That’s mainly because of the stratum corneum—the outermost layer of the skin which is made up of dead, lipid-rich cells that are tough for most substances to pass through.To tackle this challenge, scientists are turning to nano- and micro-scale technologies, especially tiny carriers like liposomes, niosomes, and ethosomes. These have shown promise in delivering drugs through the skin, but they still struggle to get past the stratum corneum. One exciting solution is deformable vesicles, known as transferosomes. Unlike traditional vesicles, transferosomes can squeeze through the skin’s tight barriers, making them a promising tool for the future of transdermal drug delivery1.Transdermal drug delivery systems are medications applied directly to the skin often as patches or creams that release drugs at a controlled rate into the bloodstream through intact skin2.
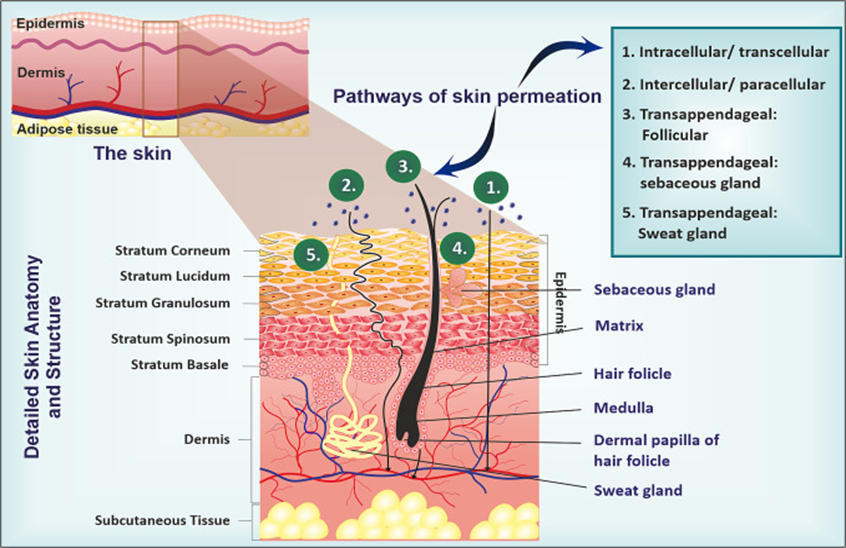
Fig:1 Detailed skin anatomy and structure
Transdermal drug delivery where medication is absorbed through the skin into the bloodstream offers a convenient option for treating various health conditions. Today, it's used to manage a wide range of diseases, including skin cancer, female sexual dysfunction, post-menopausal bone loss, urinary incontinence, depression, anxiety, ADHD, as well as cardiovascular issues, Parkinson’s, and Alzheimer’s disease. However, despite its advantages, the skin’s outermost layer the stratum corneum acts as a strong barrier, making it difficult for many drugs to pass through. This limits the broader use of transdermal delivery for other medications3.The stratum corneum the outermost layer of the skin acts as a protective shield, blocking harmful substances from entering the body. However, this same barrier also makes it difficult for medications to pass through the skin during transdermal drug delivery4. Most small, highly lipophilic drugs can naturally pass through the skin’s outer barrier, the stratum corneum. However, this makes transdermal delivery unsuitable for most hydrophilic drugs, which struggle to penetrate this protective layer. Even if a drug does manage to get through, it can be broken down by enzymes in the epidermis before it reaches circulation, lowering its bioavailability. In addition, delays in the onset of action and the lack of consistent pharmacokinetic control can make transdermal delivery less dependable5.
Transdermal Drug Delivery Systems (TDDS) are designed to overcome the skin’s natural barrier for effective drug transport. The stratum corneum (SC), with its dense and compact structure, makes this process challenging by restricting drug penetration. Traditional liposomes are limited in this regard, as they mainly remain in the upper skin layers and serve as local reservoirs. Transferosomes, however, are more advanced. By incorporating edge activators (EA), their bilayer becomes more flexible and deformable, allowing them to squeeze through the skin’s tight structure and reach deeper layers. These edge activators not only enhance flexibility but also act as permeation enhancers by disrupting the SC’s lipid arrangement, further supporting drug passage. As a result, transferosomes hold great promise for delivering drugs into deeper layers of the skin at higher concentrations6.
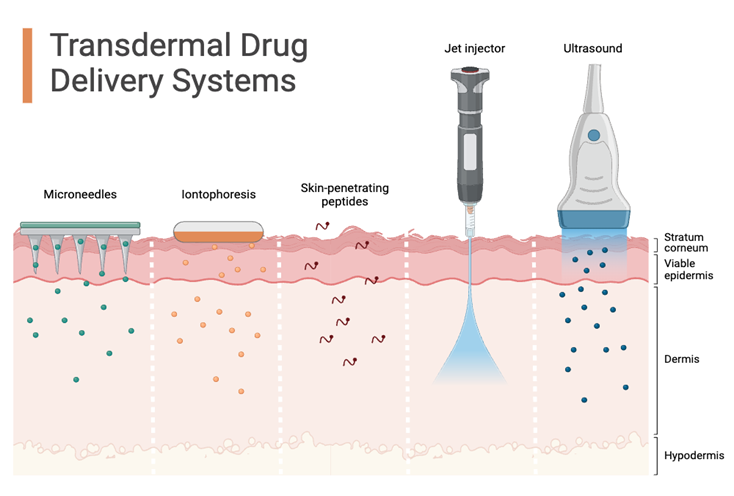
Fig:2 Transdermal drug delivery system
TRANSFEROSOMES
The term transferosomes literally means “carrying body.” It is derived from the Latin word transferre, meaning “to carry across,” and the Greek word soma, meaning “body”7.Gregor Cevc proposed the concept of transferosomes in 19918.Transfersomes are made up of phospholipids along with an edge activator (EA), which acts as a membrane-softening agent. Common examples include Tween 80, Span 80, and sodium cholate. The presence of the EA gives transfersomes their ultra-deformable nature, allowing them to pass more easily through the skin barrie9.A transferosome is a synthetic vesicle designed to mimic natural cell vesicles and is commonly used to deliver genetic material or drugs into cells. It is primarily composed of a naturally occurring amphipathic molecule, such as phosphatidylcholine, which has the ability to self-assemble into vesicles. To enhance its properties, at least one bilayer softener typically a biocompatible surfactant is incorporated. This gives the transferosome its unique structure: an aqueous core surrounded by a highly flexible and adaptable lipid bilayer. While its basic structure resembles that of a conventional liposome, the key difference lies in its “softened” bilayer membrane, which is more deformable and porous, enabling better penetration and delivery10.
Structure and composition of transferosomes:
Transferosomes are vesicles made of phospholipids, such as phosphatidylcholine, which naturally arrange into lipid bilayers in aqueous environments. To enhance their flexibility, edge activators like sodium cholate, Tween 20, or Span 80 are added, making the bilayer more elastic and fluid. This unique deformability allows transferosomes to pass through pores even smaller than their own size, efficiently squeezing through constrictions as narrow as 5–10 μm while maintaining their structural integrity. Their stress-responsive and adaptable nature makes them highly effective carriers. In addition, components like dipotassium glycyrrhizinate further improve their stability and performance. Because of their biodegradability, biocompatibility, and resistance to metabolic breakdown, transferosomes are especially promising for delivering drugs into and across the skin, achieving high efficiency depending on the route of administration and application11,12.

Fig. 3: DIAGRAM OF TRANSFEROSOME
Transferosomes are specialized vesicular carriers made of phospholipids and surfactants, designed to encapsulate both hydrophilic and hydrophobic drugs. Their unique structure contains both hydrophilic and hydrophobic regions, allowing them to accommodate a wide range of therapeutic compounds with different solubility profiles. What makes transferosomes particularly effective is their ability to deform and squeeze through constrictions that are five to ten times smaller than their own size, all while remaining intact. They can be prepared using several techniques, such as thin-film hydration, reverse-phase evaporation, or ether injection methods13.
Transferosomes are specially designed vesicles that overcome many of the limitations of conventional drug delivery systems. Made from phospholipids and surfactants, they are capable of encapsulating both hydrophilic and hydrophobic drugs. Their membranes are highly deformable and self-optimizing, meaning they can adapt to environmental stress, locally and reversibly altering their composition. This flexibility allows them to squeeze through narrow pores without significant loss, enabling intact vesicles to penetrate the skin more efficiently. Thanks to their dual hydrophilic–hydrophobic structure, transferosomes can carry drugs with a wide range of solubility profiles, including both low- and high-molecular-weight compounds. The earliest formulations typically contained soya phosphatidylcholine, sodium cholate, and a small amount of ethanol. Over time, transferosomes have been explored for delivering diverse classes of drugs, such as anti-inflammatory agents, antibiotics, antifungals, and anticancer drugs, making them a highly versatile transdermal delivery system13-16.
Table 1: Composition of transferosomes
|
Sr. No |
class |
Example |
Uses |
|
1 |
Phospholipids |
Soya phosphatidyl choline, Egg phosphatidyl choline, Dipalmitoyl phosphatidyl choline |
Vesicles forming compounds |
|
2 |
Surfactant |
Sodium cholate, sodium deoxycholate, tween 80, tween 20, span 20, span 80 |
For providing flexibility |
|
3 |
Alcohol/ Solvents |
Ethanol, methanol, isopropyl alcohol, chloroform |
As a solvent |
|
4 |
Buffering agents |
Saline phosphate buffer (pH 6.4), phosphate buffer (pH 7.4) |
As a hydrating medium |
|
5 |
Dye |
Rhodamine-123, Rhodamine-DHPE, Fluorescein-DHPE, Nile-red |
For confocal scanning Laser microscopy(CSLM) |
TRANSFEROSOMES V/S OTHER CARRIER SYSTEM
Transferosomes, niosomes, proniosomes, liposomes, ethosomes, and electrosomes are different types of vesicular drug delivery systems that have attracted significant interest in nanotechnology-based drug delivery17-20.Among these systems, transferosomes stand out as a promising option for transdermal drug delivery. Their unique properties allow them to penetrate skin pores, encapsulate both hydrophilic and lipophilic drugs, and extend the drug’s presence in systemic circulation. In addition, they can target specific organs and tissues while reducing toxicity and improving overall bioavailability18,19.
Transferosomes are flexible, deformable vesicles made up of phospholipids, surfactants, and water, designed to enhance transdermal drug delivery. Their performance is typically evaluated through parameters such as entrapment efficiency, drug content, in-vitro release, and degree of deformability, among others19.Proniosomes are dry, free-flowing formulations in which a water-soluble carrier is coated with nonionic surfactants. Upon hydration, they readily convert into niosomes. This approach helps overcome the stability issues often associated with liposomes and niosomes, while also improving the solubility, bioavailability, and absorption of various drugs17. Niosomes are small lamellar vesicles formed by hydrating a mixture of cholesterol and non-ionic surfactants, typically from the alkyl or dialkyl polyglycerol ether class, in an aqueous environment. They can serve as effective carriers for both lipophilic and amphiphilic drugs20.
Advantages
Disadvantages
MECHANISM OF TRANSFEROSOMES
Transferosomes are tiny, fat-based carriers used to deliver drugs, especially through the skin. They are made by adding water to a thin layer of lipids and then processing it at high speed to form a stable mixture of nanoparticles34.Transferosomes carry drugs into the skin’s outer layer (stratum corneum) either by passing through the cells or between them. Their main way of moving through the skin is by following an osmotic or transdermal gradient35.Colloidal particles in vesicles arrange themselves into an amphiphilic bilayer. In this structure, water-loving (hydrophilic) drugs are usually carried inside the watery core of the vesicle, while fat-loving (hydrophobic) drugs get trapped within the lipid bilayer36. On the other hand, transferosomes are highly flexible vesicles that can easily adapt their shape. Because of this deformability and their ability to interact with skin layers, they can squeeze through tiny spaces in the skin, making them ultra-flexible and capable of adjusting themselves for better drug delivery37.

Fig.4:The way transferosomes penetrate the skin works mainly through two processes: (A) they can change their shape (deform) and then return to their original form, which helps them pass through narrow gaps, and (B) they naturally move toward areas with more moisture, which supports their deeper permeation.
The drug gets incorporated into the lipid matrix of transferosomes, which helps enhance its solubility, protects it from degradation, and improves its encapsulation efficiency38.The release of a drug from transferosomes mainly follows a diffusion process, which is affected by factors like the drug’s concentration, the characteristics of the lipid matrix, and the conditions of the surrounding environment34.Transferosomes are able to penetrate deep into mucosal layers, which helps increase the drug’s bioavailability. This ability is mainly attributed to their highly elastic and flexible membranes38.Transferosomes can be used for both dermal and transdermal drug delivery. Their ability to cross the skin is mainly due to the interaction between the lipids in the transferosomes and those in the skin’s own lipid layer, which makes it easier for the drug to penetrate through the skin39.
TRANSFEROSOMES' INTERACTIONS WITH THE SKIN
Transferosomes are specially designed deformable liposomes that can pass through the stratum corneum—the outermost layer of the skin more efficiently than conventional liposomes, leading to better drug delivery40,41.They are specifically designed to improve the delivery of drugs through the skin and hold promise for use in a wide range of medical treatments40.Transferosomes are flexible, deformable vesicles made up of phospholipids, surfactants, and water, designed to enhance the delivery of drugs through the skin19.They are capable of carrying drug molecules with different solubility profiles and can serve as carriers for both low and high molecular weight drugs42.Thanks to their flexible nature, transferosomes can squeeze through narrow spaces without losing their structure. This allows them to pass through the tiny pores of the stratum corneum and reach the deeper, living layers of the skin while remaining intact43.They have been successfully used to deliver a wide range of therapeutic agents, including water-soluble drugs, large molecules, peptides, proteins, and even nucleic acids40.
FACTORS AFFECTING ON DESIGNING OF TRANSFERSOME:
Several process-related factors can influence the properties of transferosomes during the development of an optimal formulation. These factors are mainly linked to the method of preparation and are outlined as follows:
1. Effect of Phospholipids: Edge Activator: The properties of vesicles such as their size, surface charge, and ability to trap drugs are influenced by several factors. These include the concentration of surfactant, the length and number of carbon chains in the surfactant, the hydrophilic nature of the head group, the competition for space within the lipid bilayer, and the surfactant’s hydrophilic-lipophilic balance (HLB)value. In general, using a higher surfactant concentration, longer or more carbon chains, a more hydrophilic head group, and a higher HLB value tends to produce vesicles of smaller size44.
2. Effect of various solvents: Several solvents, such as methanol and ethanol, are commonly used in formulations. The choice of solvent depends on how well the ingredients are compatible with it and how easily they dissolve. For effective film formation and better stability after hydration, it is important that all components including the drug and excipients—fully dissolve in the solvent to form a clear, transparent solution. In addition, some solvents can act as penetration enhancers, helping increase the flow of the drug across biological membranes. For example, William and Barry (2004) reported that ethanol was successfully used in studies to improve the permeation of levonorgestrel, hydrocortisone, 5-fluorouracil, and estradiol through rat skin45.
3. Impact of different edge activators (surface active agents): Transfersomes are highly optimized, ultra-flexible lipid vesicles with the unique ability to quickly change shape under external pressure. This flexibility allows them to pass through skin pores that are much smaller than the vesicles themselves. The use of specific edge activators, at suitable concentrations, is essential for maximizing this membrane deformability. As a result, transfersomes act not only as efficient drug carriers but also as enhancers of drug permeation46.
4. Effect of hydration medium: The choice of hydration medium—whether phosphate buffer (pH 6.5–7) or water is critical for achieving the right balance in formulation properties, biological compatibility, and drug delivery. Maintaining the proper pH ensures that the drug stays in its unionized form, which improves its entrapment in the transfersomes and enhances its ability to pass through cell membranes. This is especially important because the lipid bilayer of transfersomes closely resembles the phospholipid layer of cell membranes, allowing efficient intracellular transport of the drug45.
THEORIES AND APPROXIMATIONS IN MICELLE AND VESICLE FORMATIONS
Micelle formation
Law of mass action.
We examine an aqueous solution of neutral amphiphilic molecules (i.e., non-ionic surfactants), each of which has a single alkyl chain as its hydrophobic tail. Amphiphiles can typically form aggregates of various sizes and shapes. We will assume that each micelle is spherical and ignore the effects of variations in its size and shape. As a result, we picture each surfactant molecule as being either a monomer or a component of a spherical n-mer. We denote the number densities of the monomers and n-mers by ρ1 and ρn, respectively, so that the total surfactant concentration is given by:
ρ = ρ1 + n ρn …..(1)
The concentrations of monomers and micelles are related by the law of mass action
ρn a3 = (ρ1a3) exp (–β? G) ….(2)
The driving force behind the assembly is the free energy of the n-mer, fn, relative to that of n monomers, nf1, where β is the inverse temperature (i.e., β-1 = kB T), and 'a' is a microscopic length that specifies the standard state convention. The diameter of a surfactant molecule is considered to be about 'a'.
ΔG = fn – nf1 …..(3)
At high values of n, equation 3 suggests that there is a threshold concentration of surfactant molecules, ρcmc, at which the density of aggregates becomes significant. The site of this crossover is nearly independent of the precise definition of the threshold, provided that it makes physical sense, because the transition is abrupt. To be precise, the errors are on the order of n-1 ln n.
In βcmc a3 = β ?G In* …..(4)
ΔG/n, the driving force per surfactant, is a function of n, and its value must be determined at the most likely aggregation number, n*. This figure represents the value of n that minimizes ΔG/n.
Driving force
The contributions to ?G can be found in three steps.
Water will occupy the area that a micelle vacates. If the surface area is at least 1 nm2, then the free energy required to produce this cavity is
?G1 = δA …..(5)
Where? The water vapor surface tension is represented by δ, and the surface area of the hole is represented by A. Additionally, creating a cavity in a liquid typically involves pressure volume effort. Pressure is low enough under normal conditions that this contribution is insignificant for holes having diameters less than 5 nm. We will only take sizes into account that fall within this range.
Consider the scenario where every hydrophobic tail of a surfactant is separated from its corresponding hydrophilic head and the hydrophobic tail is shifted from water into the micelle's core. To occupy the space created in the first step, a total of n tails must be relocated. Therefore, one component of the energy needed to occupy that space is represented as –nΔμ, where –Δμ denotes the change in free energy associated with moving the hydrophobic tail (for instance, an alkane chain) from the aqueous environment into the hydrophobic oily core. Furthermore, an additional aspect of the energy required to occupy the space stems from interfacial contributions resulting from Vander Waals forces acting between the oil and water. These forces lead to a reduction in the oil-water surface tension, γo/w, compared to the surface tension between water and vapor, γ. Consequently, the energy necessary for filling the cavity is
ΔG2 = –nΔμ – ΔγA ….(6)
where: Δγ = γ – γow .
Δμ is approximately equal to the transfer free energy for transferring the associated alkenes chain from oil into water because the inside of a micelle is extremely dense and resembles a hydrocarbon liquid. However, it is somewhat lower than this number because the environment of an alkane chain in a micelle interior is more confined than that in bulk oil. To the extent that the micelle is spherical, A = 4πL2, where L is the micelle radius. Since the interior is densely packed, L is given by A = 4πL3/3 = nδa2, where ä is the mean length over which a polar head group is separated from an alkyl group within a surfactant molecule. These considerations lead to the following conclusions:
ΔG1 + ΔG2 = –nΔμ + μgn2/3 …..(7)
g = (36π)1/3(γow a2) » 4.8 X(γow a2)(γ/a)2/3
The free energy for nucleating oil clusters in water is essentially the right-hand side of the equation. It is the hydrophobic force that the Lum-Chandler-Weeks hypothesis proposes. The volume of hydrophobic units is proportional to the first term. The area of the interface is proportional to the second term. The first term is large in n and predominates for large values of n. As a result, macroscopic clusters would form if just ΔG1 and ΔG2 were important because the driving force would increase indefinitely47.
The hydrophilic head groups are reconnected to the hydrophobic tails in the last step, placing them at the water-oil interface to preserve positive solvation energy. This positioning must be done while simultaneously enforcing the connection between heads and tails and maintaining the densely packed interior. These conditions lead to an entropic cost that rises super-extensively with aggregate size. The electrostatic analogy of stoichiometric constraints can be used to conveniently calculate the form of this third contribution to the driving force. The result is:
ΔG3 = hn5/3/β …..(8)
Where: h =34π23 9649aδ43 ? 0.75 Xaδ43

When using this comparison, it is important to remember that the volume of the micelle is mostly made up of the tightly packed alkyl chains.
Micelle size and critical micelle concentration
Combining the three contributions discussed above gives the driving force in units of kB T.
βΔG ≈ nβΔμ + βgn2/3 + hn5/3 …..(9)
Minimization of ΔG/n therefore gives:
N* ≈?g2h=49π48βγδ2
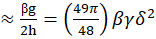
With this aggregation number:
ln ρcmc a3 = c(βγow a2)2/3 – βΔμ ….(11)
Where: c =58324913 ≈4.9

The thermodynamic cycle of micelle formation is shown in Figure 1. A straightforward geometric model, which defines a dimensionless critical packing parameter (CPP) to represent the relative bulkiness of the hydrophobic and hydrophilic components of an amphiphile, may be used to correlate the structure of the amphiphile with its phase behavior. As the CPP rises from a low to a high value, the amphiphile switches its preferred phase structure from direct structures through lamellar structure to reverse structures, changing from hydrophilic to hydrophobic. The foundation for the molecular design of amphiphiles is this model.
Molecules on the surface feels curvature when the size is small and molecules experience stress.
Fig.5: Thermodynamic cycle of micelle formation.
The process of assembling n separated am phiphiles (a) to a micelle (d) can be performed in three steps: 1 – creating a cavity in the solvent (light gray) (b), 2 – transferring the hydrophobic chains (dark gray) from the aqueous solution into the cavity (c), 3 – distributing the polar units (gray) over the surface of the cavity and recon necting them to the hydrophobic groups (d)
The stress gets significant role when the size is below 100nm. For bilayer formation,the molecules must be amphiphilic.Typical geometry for a vesicle formation is:
P=V/ a0 Ic …...(12)
where:
A pictorial representation of geometry and variants of packing arrangements are shown in Figure 2 and 3 respectively.
Vesicle formation
As previously stated, tiny, consistently sized egg lecithin vesicles were made. At this moment, any solute that needed to be trapped was introduced to the vesicle solution, and the vesicles were brought to 20 mM phospholipids. After that, the mixture was heated to 25°C, and 250 mM sodium deoxycholate was quickly added and mixed to create a last mixture with a 1:2 ratio of deoxycholate to phospholipids. The large vesicles started to form almost immediately, as shown by the rise in the solution's light scattering, which went from being nearly transparent for the small vesicles to being transparently opalescent for the large vesicles. The formation of the vesicles was finished in between 5 and 10 minutes.
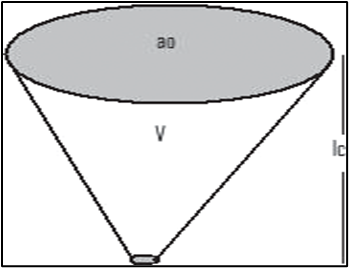
Fig.6: Representation of typical geometry for a vesicle formation, P = V/ a0 Ic
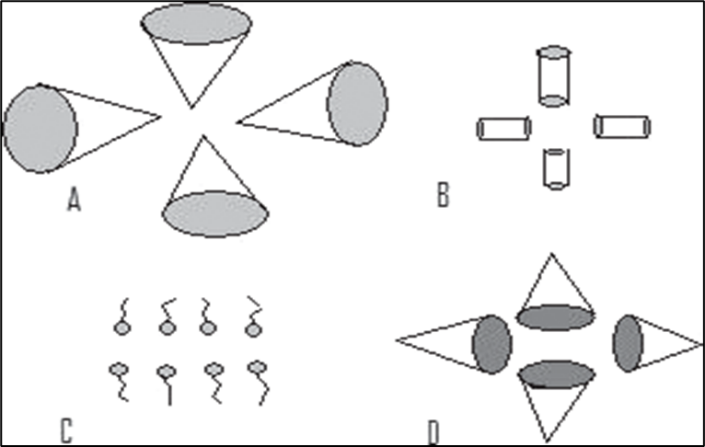
Fig.7: A. P = 1/3, micelle formation, B. P = 1/2, cylindrical micelle, C. P = 1, planar bilayer, D. P ? 1, inverted micelle
The majority of the detergent (96-98%) was then eliminated by passing the sample over 60 volumes of Sephadex G-25 (medium porosity) at 250°C (vesicles may potentially be produced at 40°C and need 15-30 minutes). It was discovered that the remaining deoxycholate could be easily removed by a second gel filtration column of 20–30 volumes, which may indicate that the detergent had become trapped inside the vesicle and had re-equilibrated with the outside volume after the bulk of the de- tergent had been removed. The final product had fewer than one deoxycholate molecule for every thousand phospholipid molecules. The vesicles are now stable for several weeks, as seen by the absence of change in turbidity or size when viewed under an electron microscope. It seems that the chemical stability of the unsaturated phopholipid component restricts the stability.
Egg lecithin (20 μmol) was dried into a thin film using chloroform in a 15 ml Cortex tube while applying reduced pressure. A tube was swirled after adding one milliliter of buffer with 10 mM sodium deoxycholate and the solute that needed to be trapped in the vesicles. The mixture was briefly stirred using a Bransonic12 sonifier in a bath at 25°C while nitrogen gas was used. After 1 to 2 minutes of sound waves being used, the solution changed from a cloudy, milky color to a clear, shiny look. Sonication for an additional 15 minutes did not change how the solution looked, and it did not make the cloudiness go down any more. At this stage, they took out deoxycholate.
Vesicles are grouped into three types:
The standard steps for making vesicular preparations are:
The organic solvent method helps create multilamellar vesicles (MLVs) by adding water to them. MLV sextrusion through 0. To create large unilamellar vesicles (LUVs), we use a 1 μm polycarbonate filter along with a freeze-thaw method, reverse phase evaporation, and steps to remove detergent. Sonication is a method used to create small unilamellar vesicles (SUVs).
The direct hydration process has some benefits, including being quick. Its drawbacks are a small amount of trapped volume, low efficiency in trapping, and uneven distribution of the solute.
The benefit of using an organic solvent for hydration is that it captures substances very effectively. However, it has some downsides, such as being complicated to use and being restricted by how well lipids can dissolve in the organic phase.
The process of removing detergent has some benefits, like being able to restore proteins and having a large volume of trapped material. However, it also has drawbacks, including the challenge of completely getting rid of the detergents and a low efficiency in trapping48
SCOPE OF TRANSFEROSOMES:
Transferosomes(TFs) have a special ability to change their shape and size on their own. This allows them to pass through the small gaps in healthy skin quickly and easily. The difference in water content between the dry outer layer of the skin, which has about 15% water, and the wetter inner layer, which has around 75% water, creates a pressure that helps move tiny substances called TFs through the skin. It is a very flexble and stress-sensitive group of things that work together. When the carrier is put on the skin, it looks for and uses tiny openings, called pores, between the skin cells. It stretches these pores wide enough to allow the whole vesicle and its medicine to pass through. It can change shape a lot to do this while still keeping its structure intact. The way the local structure and shape of the bilayer work together allows the vesicle to control itself and improve its own performance. This allows the Transferosome to easily move across different transport barriers. Transfersome can pass through the outer layer of skin either by going between the cells or by moving through the cells themselves49.
TFs are more flexible than regular liposomes, making them a new type of carrier that works well for delivering medicine through the skin50. TFs keep the drug inside safe from being broken down by the body. They function like a storage place, slowly and gradually releasing their contents. These tiny carriers have an edge activator (EA) like sodium deoxycholate, span 80, or tween 80. This feature allows them to adapt to stress, making it easier for them to fit through the small openings in the outer layer of the skin. Transferosomes are tiny carriers that can easily build up in the loose tissue around joints, allowing for better targeting in those areas51.
TFs are special types of delivery systems used on skin that does not block air. They release medication in a way that enhances its effectiveness while also reducing the chances of patients not following their treatment plans. Additionally, they can stop the liver from breaking down the drug before it enters the bloodstream, which leads to improved effectiveness in how the body processes the medication. Ultradeformable vesicles can easily take in herbal extracts and synthetic ingredients, delivering them intact to the targeted areas without any leakage of the drug. These vesicular carriers have become important tools for treating different skin conditions. They help overcome barriers that limit how well treatments can penetrate the skin, which improves their effectiveness and leads to significant health benefits. These carriers also offered new ways to deliver active ingredients through the skin in an effective and interesting way. Confocal scanning laser microscopy has been utilized to study how transferosomes enter cells. Transferosomal drug delivery is commonly used to effectively treat different diseases. It offers improved compatibility with the body, better absorption, lower costs, and increases patient satisfaction. This review highlights the development and advantages of transferosomes, a type of vesicle, in improving treatment effectiveness and usefulness. This paper discusses key features, benefits, preparation techniques, and important uses of transferosomes. Sure! Please provide the text you'd like me to paraphrase52.
SALIENT FEATURES OF TRANSFERSOMES :
Salient Features of Transferosomes: These are made up of natural phospholipids like liposome therefore these are biodegradable and biocompatible
DESIGNING OF TRANSFEROSOMES:
Materials commonly used for the preparation of transfersomes
Transferosomes are innovative, ultra deformable vesicular systems designed for advanced drug delivery. They are mainly made up of three key ingredients: phospholipids, which form the structural base of the vesicle; surfactants or edge activators, which enhance the flexibility of the vesicle membrane; and water, which acts as the medium in which these vesicles are formed. This unique combination allows transferosomes to squeeze through narrow spaces in the body, making them especially useful for delivering drugs through the skin or other biological barriers.54,55. Ethanol may sometimes be incorporated into the transferosomalformulation56.
Phospholipids, typically making up more than 70% of the formulation, are a key component in the preparation of transferosomes. Commonly used types include phosphatidylcholine (sourced from eggs or soybeans), as well as synthetic variants like dipalmitoyl phosphatidylcholine, dimyristoyl phosphatidylcholine, distearoyl phosphatidylcholine, and dipalmitoyl phosphatidylglycerol. These phospholipids help form the flexible bilayer structure that gives transferosomes their unique deformability. 54,56. Various edge activators (ranging from 10 to 25%)such as surfactant(e.g., Tween R 20, TweenR 60, TweenR 80, SpanR 60, SpanR 65 and SpanR 80) or bile salt (e.g., sodium cholate and sodium deoxycholate) are generally incorporated into the TFs.
Phosphate-buffered saline (PBS), typically at pH 6.4 or 7.4, is also commonly used as a hydrating medium in transferosome formulations, helping to maintain a stable pH environment during preparation58.
Ethanol can also be included in transferosome formulations, either in small amounts (less than 10%) or in higher concentrations (10–50%), similar to what's used in ethosomes. Its presence can enhance membrane flexibility and improve drug penetration through the skin.56.
Dyes are often used in transferosome studies to help visualize and track vesicle penetration through the skin, especially during confocal scanning microscopy analysis59.
The movement of transferosomes across the skin is driven by a natural water gradient. The skin’s outer surface is relatively dry, containing about 15% water, while the deeper layers of the epidermis have much more—around 75% water. This difference in water content creates an osmotic pressure that helps pull the transferosomes through the skin, making drug delivery more effective58,54,56,60.
When transferosomes are applied to the skin, they can easily lose water and dry out. To prevent this, they use their unique flexibility—thanks to a property called elasto-mechanics—to squeeze through the tiny gaps in the outer layer of the skin (the stratum corneum). Once they reach the deeper, more hydrated layers, they absorb water and return to their original shape, allowing them to effectively deliver their cargo56.
METHOD OF PREPARATION
Preparing transferosomes involves two main steps. First, a thin film is created and then hydrated to form vesicles, which are then sized down using sonication. Second, these sonicated vesicles are further refined and made uniform by passing them through a polycarbonate membrane using a process called extrusion.
PREPARATION OF TRANSFEROSOMES
1. THIN-FILM HYDRATION METHOD:
The phospholipids and edge activators, which form the vesicles, are dissolved together in a round-bottom flask using a mix of volatile organic solvents—usually a specific ratio of chloroform and methanol. This step also allows for the lipophilic drug to be incorporated into the mixture. Then, using a rotary vacuum evaporator, the solvents are gently removed under reduced pressure and at a temperature above the lipid transition point, leaving behind a thin, even layer of lipids62-65.
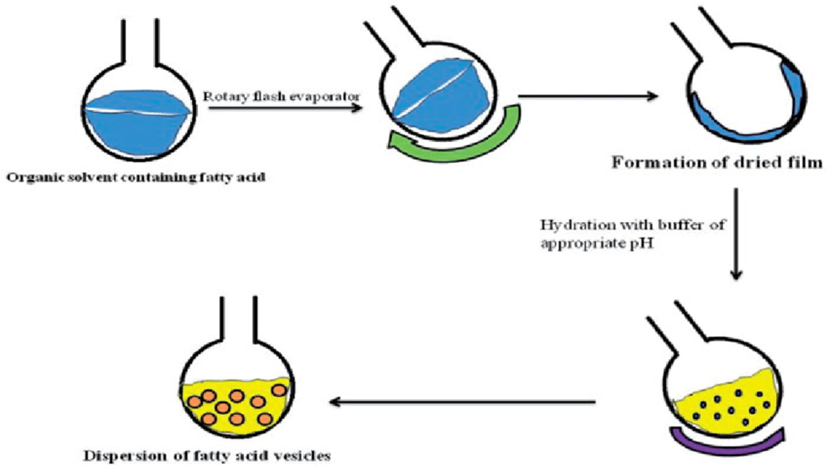
Figure 8: Preparation of transerosomes
Thin-film hydration method of preparation of fatty acid vesicles. Rotation of the round-bottom flask is done by rotary flash evaporator but at the lab scale hand shaking method is also adopted80.
Any remaining traces of solvent were removed by keeping the mixture under vacuum overnight. After that, the thin layer of deposited lipids was hydrated using a buffer solution (pH 6.5) while rotating the flask at 60 rpm at a temperature suitable for the formulation59.
This is also the stage where hydrophilic drugs can be added and incorporated into the formulation62-65.
To create smaller vesicles, the large multilamellar vesicles (LMVs) are sonicated either at room temperature or at 50°C for 30 minutes using a B-12 FTZ bath sonicator. Alternatively, they can be probe-sonicated at 40°C for 30 minutes using a titanium micro tip sonicator (Heat Systems W 380)59.
After sonication, the vesicles are further refined and made uniform by passing them through a stack of polycarbonate membranes with pore sizes ranging from 200 to 100 nanometers, using a process called extrusion62-65.
Out of the different methods available, the thin-film hydration technique using rotary evaporation is the most popular for developing transferosomes at the lab scale. This is because it’s simple, quick, cost-effective, and safe. It’s especially suitable for heat-sensitive materials and doesn’t require any complex or expensive equipment 56.
In the vortexing-sonication method, phosphatidylcholine, edge activators, and the therapeutic molecule are mixed in an appropriate phosphate buffer and vortexed to form a milky suspension. This mixture is then sonicated at room temperature, followed by extrusion through 450 nm and 220 nm membranes to produce transferosomes.
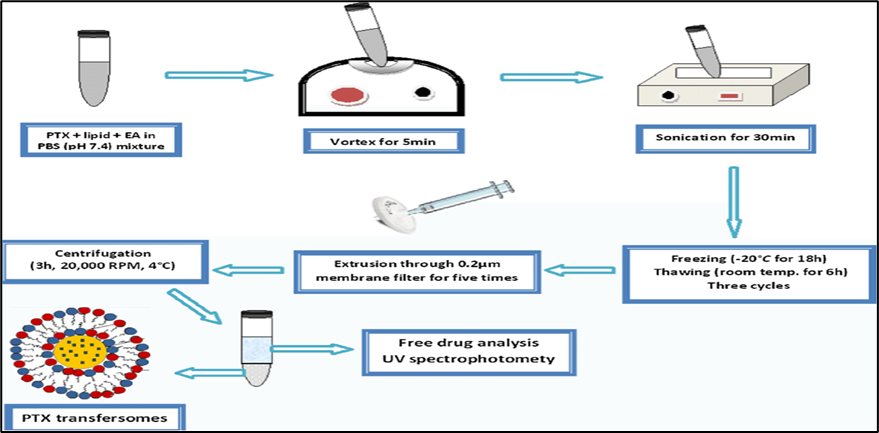
Fig:8 Preparation procedure of transfersomes formulations 81
For another formulation, RES, soya lecithin, and a surfactant are dissolved in a 2:1 mixture of chloroform and methanol. The solution is then evaporated to dryness and hydrated using a simulated nasal fluid (SNF) at pH 5.5. The resulting vesicles are size-reduced using a probesonicator, filtered through a 0.2 µm membrane, and finally cooled and stored at 4°C66-68.
This method of preparing transferosomes is widely recognized for its efficiency and effectiveness in delivering therapeutic agents directly to the target site69-70.
The modified handshaking method is based on the same core principles as the traditional rotary evaporation-sonication technique. In this approach, the organic solvent, lipophilic drug, phospholipids, and edge activator are combined in a round-bottom flask and mixed until a clear, transparent solution is formed, ensuring complete dissolution of all components. Instead of using a rotary vacuum evaporator, the organic solvent is gently evaporated by hand-shaking the mixture above the lipid transition temperature (around 43°C)71.During this process, a thin lipid film gradually forms along the inner wall of the flask as the solvent evaporates overnight. Once the film is fully dried, it is rehydrated using an appropriate buffer solution containing the hydrophilic drug. This step is carried out with gentle shaking at a temperature above the lipid’s phase transition point to ensure proper hydration. The method closely resembles the traditional thin film hydration technique, where lipophilic drug, organic solvent, edge activator, and phospholipids are combined in a flask. The goal remains consistent: to achieve complete dissolution and a clear, uniform solution. As the solvent is slowly removed through handshaking, a thin lipid layer is deposited on the flask’s inner surface.66,67,68.
This method is especially effective for targeted drug delivery, as the vesicles can penetrate the skin's stratum corneum and reach the systemic circulation. Overall, the transferosome preparation technique holds great promise in advancing drug delivery research. Figure 2 provides an illustration of the Rotary Film Evaporation Method, also known as the Modified Hand Shaking Method, used for formulating transferosomes69-70.
Transferosomes are prepared by blending an ethanolic solution of soybean phosphatidylcholine with a suitable amount of an edge activator, such as sodium cholate66,67,68.The resulting suspension is then mixed with a Triethanolamine-HCl buffer to achieve the desired total lipid concentration. This mixture undergoes sonication, followed by two to three freeze-thaw cycles to enhance vesicle formation. Once processed, the vesicles are reduced to the required size and analyzed using photon correlation spectroscopy. To ensure sterility, the preparation is filtered through a 0.2 μmmicroporous filter. Finally, the size of the vesicles is confirmed using Dynamic Light Scattering (DLS) 72,73,74.
In this process, phospholipids, an edge activator, and the lipophilic drug are first dissolved in an organic solvent. The solvent is then removed using a rotary evaporator under reduced pressure at the appropriate temperature, and any remaining traces are eliminated under vacuum. The resulting thin lipid film is hydrated with a suitable buffer solution, allowing for the incorporation of a hydrophilic drug. This step is followed by centrifugation at room temperature, leading to the formation of vesicles, which are allowed to swell naturally.
At the same time, a parallel procedure is carried out where phospholipids, surfactants, and the drug are dissolved in alcohol. The solvent is again removed by rotary evaporation under reduced pressure at 40°C, with any residual traces removed under vacuum. The resulting lipid film is hydrated using a buffer solution and centrifuged at 60 rpm for one hour at room temperature. The vesicles from this process are then allowed to swell for an additional two hours.
Finally, the multilamellar vesicles obtained from both methods are sonicated at room temperature to further reduce their size and improve uniformity66,67,68.
In this method, phospholipids and an edge activator are first dissolved in a mixture of organic solvents—typically diethyl ether and chloroform—in a round-bottom flask, allowing the incorporation of a lipophilic drug. After forming a thin lipid film through rotary evaporation, the lipids are redissolved in an organic phase mainly composed of isopropyl ether and/or diethyl ether. An aqueous solution containing surfactants (edge activators) is then added, creating a two-phase system that supports the incorporation of hydrophilic drugs. The mixture is sonicated using a bath sonicator to form a uniform water-in-oil (w/o) emulsion. Gradual evaporation of the organic solvent using a rotary evaporator transforms the emulsion into a viscous gel, which eventually forms a vesicular suspension.
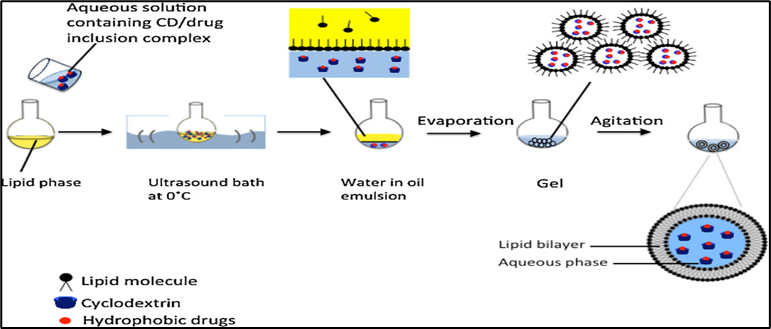
Fig. 9. Reverse phase evaporation method82.
In an alternative approach, lipids are dissolved in an organic solvent within a round-bottom flask. An aqueous phase containing edge activators is then added under nitrogen purging. Depending on its solubility, the drug is incorporated at this stage. Sonication is used to produce a homogeneous dispersion, and the organic solvent is removed under reduced pressure. As the solvent evaporates, a viscous gel forms and later transitions into vesicles. Any non-encapsulated drug or residual solvent is removed through dialysis or centrifugation to ensure purity and stability75-76.
To begin, phospholipids, an edge activator, and the drug are continuously mixed in distilled water. This mixture is then treated using ultrasonic shaking to promote uniformity, followed by high-pressure homogenization to further refine the particle size. The resulting transferosomes are then stored under appropriate conditions.
In the ethanol injection method, phospholipids, the edge activator, and a lipophilic drug are dissolved in ethanol using a magnetic stirrer until a clear, transparent solution is formed—this serves as the organic phase. Meanwhile, the aqueous phase is prepared by dissolving water-miscible components in a phosphate buffer, allowing for the incorporation of hydrophilic drugs at this stage. Both phases are then heated to around 45–50°C. The ethanolic solution is slowly added to the aqueous phase while stirring continuously. To remove the ethanol, the resulting dispersion is subjected to vacuum evaporation and then sonicated to reduce the particle size and ensure a uniform formulation 61.
To obtain the transfersomal formulation, a method involving alternate cycles of freezing and heating is employed. The multilamellar vesicles (MLV) are exposed to very low temperatures, by dipping for 30 s at −30°C(nitrogen bath), followed by high temperature exposure in a water bath. The steps are repeated several times to produce the desired transferosomes79.
The organic phase is prepared by dissolving phospholipids, an edge activator, and the lipophilic drug in ethanol, using magnetic stirring until a clear solution is achieved. Meanwhile, the aqueous phase is made by dissolving water-soluble substances, including the hydrophilic drug if needed, in phosphate buffer. Both solutions are then gently heated to 45–50°C. Next, the ethanolic phospholipid solution is slowly injected drop by drop into the aqueous phase while stirring continuously. To remove the ethanol, the resulting mixture is placed in a vacuum evaporator and then sonicated to reduce particle size, producing a uniform and stable dispersion.77-78.

Fig:10 Ethanol injection method82
CHARACTERIZATION AND EVALUATION OF TRANSFERSOMES
Transferosomes share similar characterization features with liposomes, niosomes, and micelles. Several established methods are available to evaluate their properties, including vesicle shape and size, size distribution, polydispersity index, zeta potential, vesicle count per cubic millimeter, entrapment efficiency, degree of deformability, and skin permeability measurements83-85.
These evaluation parameters play a key role in assessing the effectiveness of transferosomes, including their drug-loading capacity, drug-release profile, and overall therapeutic potential as ultraflexible liposomal formulations. Evaluating transferosomes is essential to understand the relationship between their key components and the processing factors involved in developing optimized nanometric formulations86,87.
A detailed explanation of each of the characterization methods is provided below.
Vesicle size is a critical parameter in the preparation of transferosomes, as well as in batch-to-batch comparisons and scale-up processes. Changes in vesicle size during storage are an important indicator of the formulation’s physical stability. Vesicle size also affects the encapsulation efficiency of drug compounds: a high lipid-to-core ratio is preferred for lipophilic and amphiphilic drugs, whereas a larger aqueous core is more suitable for encapsulating hydrophilic compounds88-90.
Transferosomes can be visualized using transmission electron microscopy (TEM), while their vesicle size and size distribution are typically measured by dynamic light scattering (DLS). Vesicle diameter can also be determined using photon correlation spectroscopy. For these analyses, samples are prepared in distilled water, filtered through a 0.2?µm membrane, diluted with filtered saline, and then measured using either photon correlation spectroscopy or DLS91.Zeta potential is measured to assess the stability of transferosomes, while microscopy provides detailed visualization. This comprehensive approach ensures that transferosomes are suitable for effective drug encapsulation90.
Transferosome vesicles can be visualized using transmission electron microscopy (TEM) at an accelerating voltage of 100?kV. They can also be observed without sonication through phase contrast microscopy using an optical microscope. Additionally, dynamic light scattering (DLS) can be employed to determine vesicle shape92.
This parameter is essential for optimizing the composition and other processing variables. Transferosome formulations (without sonication) can be diluted fivefold with a 0.9% sodium chloride solution92.Further studies utilize a hemocytometer along with an optical microscope, enabling the observation of transferosomes with vesicle sizes above 100?nm. This analysis provides valuable insights for refining both the composition and process parameters of the formulations85,93.
The number of transferosomes in 80 small squares is counted and calculated using the following formula:
Total no of transferosomes per cubic mm=Total number of Transferosomes counted×Dilution factor×4000Total number of squares counted
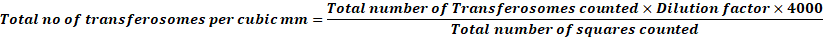
The amount of drug encapsulated is determined by breaking open the opaque vesicles. After centrifugation, the vesicles are disrupted, and a systematic approach is used to measure the drug content94.Entrapment efficiency (%EE), representing the percentage of drug encapsulated within the vesicles, is determined by separating the unentrapped drug from the transferosomes using techniques such as mini-column centrifugation83. A 1 mL sample of the transferosome dispersion was placed in an Eppendorf tube and frozen at −20 °C for 24 hours. The frozen samples were thawed at room temperature and centrifuged at 14,000 rpm for 50 minutes at 4 °C. The resulting pellet of transferosomes was resuspended in PBS (pH 6.4) and centrifuged again. This washing process was repeated twice to ensure complete removal of any unencapsulated drug. After each centrifugation, the supernatant was collected for measurement of free drug content. Drug concentration was determined spectrophotometrically at 306 nm, using PBS (pH 6.4) as the blank. Each value represents the mean (±SD) of three replicates. The entrapment efficiency (%EE) was then calculated as the percentage of drug encapsulated in relation to the total drug concentration, using the following formula95:
Percentage Entrapment Efficiency (%EE) =Amount of the drug entrapped / Total amount of the drug added ) × 100
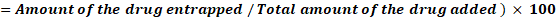
The indirect method involves diluting the supernatant with an appropriate solvent and filtering it to remove impurities. The drug concentration in the supernatant, representing the unencapsulated (free) drug, is then determined using an analytical method and calculated as follows83,96:
%Entrapment efficiency= total amount of drug added- amount of free drugtotal amoumt of drug added x 100
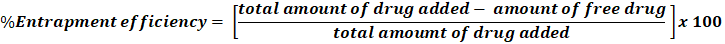
A Zetasizer is used to measure the surface charge and charge density of transferosomes97.
A nephelometer is commonly used to measure the turbidity of aqueous solutions98.
For transferosomes, permeability studies are an important and distinctive parameter for their characterization99.The percentage of drug entrapment is a critical factor, as it directly affects the permeation of transferosomal formulations. In this study, using pure water as the standard, the transferosome preparations were passed successively through microporous filters ranging from 50 to 400?nm. Particle size and distribution were monitored after each pass using dynamic light scattering (DLS), while permeability and deformability were assessed as key parameters for transferosome characterization83,90,100.
The degree of deformability or permeability is expressed using the following formula:
D = (rv/rp)×J
Where;
Before conducting costly in vivo studies, the transport efficiency of transdermal delivery systems is assessed, and factors affecting drug flux (expressed as μg/cm²/h) are identified. Experiments are performed using Franz diffusion cells with pig skin, synthetic membranes such as Strat-M®, or modified cells incorporating goat skin to simulate in vivo conditions. Synthetic membranes have been shown to correlate closely with human skin101.
A modified Franz diffusion cell with a receiver compartment volume of 50?mL and an effective diffusion area of 2.50?cm² was used for this study. In vitro drug permeation experiments were conducted using goat abdominal skin in phosphate buffer solution (pH?7.4). Fresh abdominal skin was collected from a slaughterhouse, with hairs removed and the tissue hydrated in normal saline. The underlying adipose layer was carefully removed using a cotton swab. The prepared skin was then stored in isopropyl alcohol at 0–4?°C until use102.
For the skin permeation study, the prepared skin was mounted horizontally on the receptor compartment of the Franz diffusion cell, with the stratum corneum facing upward toward the donor compartment. The effective permeation area exposed to the receptor compartment was 2.50?cm², and the receptor compartment had a volume of 50?mL. It was filled with phosphate buffer (pH?7.4), maintained at 37?±?0.5?°C, and stirred continuously at 100?RPM using a magnetic bar. The formulation, containing an equivalent of 10?mg of the drug, was applied onto the skin, and the diffusion cell was covered. At predetermined time intervals, 1?mL aliquots of the receptor medium were withdrawn and immediately replaced with an equal volume of fresh phosphate buffer to maintain sink conditions. Correction factors for each withdrawn aliquot were applied when calculating the drug release profile. Samples were then analyzed using an appropriate instrumental analytical technique103.
This study allows for the comparison of transferosomes with other vesicular systems such as liposomes and niosomes, as well as the investigation of the mechanism of transferosome penetration. The method involves incorporating a lipophilic fluorescent marker, which emits light that can be detected and used for further analysis104,89.
Conventional light and electron microscopy often face challenges with fixation, sectioning, and staining of skin samples. In many cases, the structures being examined are incompatible with these processing techniques, which can lead to misinterpretation. These issues can be minimized by using Confocal Scanning Laser Microscopy (CSLM), which allows for more accurate visualization of skin structures105.
In this technique, lipophilic fluorescent markers are incorporated into the transferosomes, and the light emitted by these markers is used for the following purposes:
Different fluorescence markers used in CSLM study are as –
Confocal microscopy offers enhanced lateral and axial resolution compared to standard light microscopy. Axial resolution is particularly important for identifying the precise location of vesicles deep within tissues. One of the main advantages of a confocal microscope is its ability to optically section thick specimens. Real-time video frames can be captured using a low-light video camera connected to a recorder, providing a live demonstration of vesicle pathways and transport behavior. Using confocal laser scanning microscopy (CLSM), it is possible to locate and study the transport of fluorescent substances with a high degree of precision across different materials92.
The tape stripping technique is a widely used method for examining the localization and distribution of substances within the stratum corneum. It provides a simple approach to reducing the barrier function of the stratum corneum by physically removing its layers. In this technique, adhesive tape is applied to the skin to remove a layer of corneocytes. In vivo, the stratum corneum is sequentially removed by repeated application of adhesive tapes to the skin surface. This method can be used to assess stratum corneum cohesion by quantifying the amount of tissue removed. Tape stripping can also be combined with techniques such as electron microscopy and FT-IR for further analysis. Several factors influence the amount of stratum corneum removed, including the tape stripping method, skin hydration, cellular cohesion, body site, and inter-individual variability106-116.
The in vitro drug release profile offers essential insights into formulation design, shedding light on the release mechanism and kinetics, and supporting a scientific approach to optimize transferosomal formulations. Typically, the in vitro release of transferosomes is evaluated in comparison with the free drug or a reference product. Numerous studies have successfully demonstrated the drug release profiles of developed transferosome formulations117.In vitro drug release studies are conducted to determine the permeation rate, including the time required to reach steady-state permeation and the flux at steady state. The information obtained from these studies is used to optimize the formulation before proceeding to more costly in vivo experiments118,119.
For in vitro drug release studies, the beaker method is commonly used. In this approach, the transferosome suspension is incubated at 32?°C using a cellophane membrane. Samples are collected at predetermined time intervals and analyzed using various analytical techniques, such as UV spectrophotometry, HPLC, or HPTLC. The free drug is separated by mini-column centrifugation, and the amount of drug released is then calculated120.
The stability of transferosome vesicles is evaluated using DLS and TEM to monitor changes in size and structure over time. Optimized formulations are stored in sealed amber vials following ICH guidelines: long-term storage at 25?±?2?°C/60?% RH?±?5?% or 30?±?2?°C/65?% RH?±?5?%, and accelerated storage at 40?±?2?°C/75?% RH?±?5?%. Refrigerated products are stored long-term at 5?±?3?°C and accelerated at 25?±?2?°C/60?% RH?±?5?%. A significant change is defined as any deviation from the established specifications121.
The initial percentage of drug entrapped in the formulation was determined, and the samples were stored in sealed glass ampoules. The ampoules were kept at 4?±?2?°C (refrigeration), 25?±?2?°C (room temperature), and 37?±?2?°C (body temperature) for at least three months. Samples were withdrawn from each ampoule at 30-day intervals to assess drug leakage. The percentage of drug loss was calculated by considering the initial drug entrapment as 100?% 102,122.
The drug content is typically analyzed using instrumental methods, most commonly a modified high-performance liquid chromatography (HPLC) technique. This setup includes equipment such as an ultraviolet detector, column oven, autosampler, pump, and a computerized system for data analysis, following the relevant pharmacopoeial method123.The choice of analytical method depends on the procedure specified for the drug in the relevant pharmacopoeia120.
The penetration capability of transferosomes is commonly assessed using fluorescence microscopy, a widely employed method for evaluating their skin permeation124.
While occlusion of the skin can enhance drug permeation in traditional topical formulations, it can be detrimental for elastic vesicles such as transferosomes. In vesicle-mediated skin delivery, hydrotaxis—the movement of water from the drier surface to the more hydrated deeper layers—serves as a key driving force. Hydrotaxis helps maintain hydration gradients and prevents water loss from the skin. Occlusion can interfere with these hydration forces by limiting water evaporation, which may reduce the permeation efficiency of elastic vesicles97.
At the end of the 24-hour permeation experiments, the skin surface was washed five times with a 1:1 mixture of ethanol and PBS (pH?7.4), followed by rinsing with water to remove any residual drug. The skin was then cut into small pieces and homogenized in the same ethanol–buffer mixture (1:1) for 6?hours at room temperature. After shaking for 5?minutes and centrifuging at 5000?rpm for 5?minutes, the drug content in the supernatant was analyzed following appropriate dilutions with phosphate buffer (pH?7.4). The results were compared to the control group using Student’s t-test97.
DSC thermograms were recorded using a Shimadzu DSC-50 differential scanning calorimeter. Analyses were performed on NYS powder, plain transferosomes, and transferosomes loaded at a 90:10?% (w/w) ratio of PL:EA. Samples of 40?µL or 1?mg were sealed in standard aluminum pans. Thermograms were obtained under a nitrogen atmosphere over a temperature range of 0–300?°C at a scan rate of 10?°C/min, using phosphate buffer (pH?7.4) as the medium95.
The surface morphology and shape of the loaded transferosomes were analyzed using transmission electron microscopy (TEM) (100?CX, Jeol, Tokyo, Japan). The transferosomes were dispersed in water, and a drop of the diluted suspension was placed on a carbon-coated grid. The sample was allowed to stand for 2?minutes to enable absorption onto the carbon membrane, and excess liquid was removed with filter paper. A drop of 2?% ammonium molybdate was then applied as a negative stain, with any excess washed off using distilled water. The prepared samples were subsequently examined under TEM to visualize the vesicles95.
Once transferosomes penetrate the outermost layers of the skin, they can enter the bloodstream via the lymphatic system and be distributed throughout the body when applied under appropriate conditions. Through transdermal delivery, transferosomes can provide drugs to body tissues accessible to subcutaneously injected liposomes. The kinetics of an epicutaneously applied agent depend on both the rate of carrier penetration and the subsequent speed of drug distribution and action. The most important factors influencing this process are:
The onset of the penetration-driving force depends on the volume of the suspension medium that must evaporate from the skin surface before a sufficiently strong transcutaneous chemical potential or water activity gradient is established; using less solvent is favorable in this regard. The rate at which carriers pass through the skin is mainly determined by the activation energy required for carrier deformation. If transferosome penetration involves occluding the application site or using an overly diluted suspension, the process can be hindered. Additionally, the elimination of carriers from the subcutaneous tissue is largely influenced by lymphatic flow, meaning that factors such as general anesthesia, which affect lymphatic circulation, can alter the rate of transcutaneous carrier transport.
Drug distribution is also influenced by the number of carriers used, as this can affect the rate of vehicle degradation or filtration within the lymph nodes. Consequently, the lag time between drug application and its appearance in the body is often long, complex, and highly dependent on the type of drug and the formulation. In optimal conditions, rapidly exchanging agents, such as local analgesics, can be detected just beneath the skin barrier within approximately 15 minutes. Slower-exchanging molecules, or those measured in the bloodstream, typically show a lag time of 2 to 6 hours, depending on the formulation details. Molecules that do not readily diffuse from their carriers, or those delivered with suboptimal carriers, generally fall into this slower category. The kinetics of vesicle penetration into and across the skin can be largely controlled by adjusting the physicochemical properties of the carrier suspension. The penetration kinetics of transferosomes through intact skin are best studied using direct biological assays, where vesicle-associated drugs exert their effects immediately beneath the skin surface.
Local analgesics are particularly useful for studying penetration kinetics. In one study, various lidocaine-loaded vesicles were applied and left to dry on intact skin, with subcutaneous injection used as a control. The animal’s pain sensitivity at the treated site was then measured over time. Neither dermally applied standard liposomes carrying lidocaine nor simple lidocaine solution produced any noticeable analgesic effect; significant pain relief was only achieved when these preparations were injected. In contrast, lidocaine-loaded transfersomes showed clear analgesic activity even when applied topically. The maximum effect was typically observed around 15 minutes after application, and a marked analgesic effect persisted for an extended period. The depth and kinetics of transfersome penetration through the skin are influenced by several factors, including drug–carrier interactions, the form and conditions of application, skin characteristics, and the applied dose99,97,125.
Table:2 The parameters and the testing methods
|
Parameter |
Method/ Equipment |
|
Zeta potential |
Electrophoretic mobility technique |
|
Vesicle size, size distribution |
Dynamic light scattering method (DLS Method) |
|
Vesicle morphology |
DLS method, Photon correlation spectroscopy and Transmission electron microscopy |
|
Number of vesicles for cubic mm |
Hemocytometer and optical microscope |
|
Entrapment efficiency |
%Entrapment efficiency = (Amount of the drug entrapped / Total amount of the drug added ) × 100 %Entrapment efficiency = [(total amount of drug added - amount of free drug)/total amoumt of drug added ] x 100 |
|
Drug content |
The HPLC method has been adapted by incorporating an ultraviolet detector, auto-sampler, column oven, pump, and computerized analysis program, tailored to the specific analytical requirements of the active pharmaceutical agent. |
|
Degree of deformability |
Microporous filter with DLS and Transmission electron microscopy |
|
Surface charge and charge density |
DLS method by Malvern Zetasizer and Thin-layer chromatography |
|
In vitro drug release |
Franz diffusion cell with cellulose membrane and Extrusion method |
|
In vivo skin permeation studies |
Franz diffusion cell |
|
In-vivo permeation ability |
Histological study used to determine the bioadhesive potential and retention of transferosomes in the skin, Confocal scanning laser microscopy (CSLM), Fluorescence microscopy |
|
Stability studies |
DLS method and transmission electron microscopy |
Application of Transfersomes
Transferosomes are lipid-based nanosystems designed for topical drug delivery. They offered significant advantages, such as improving the solubility and permeability of drugs with low bioavailability. In various studies, they were successfully used to deliver different drugs, including antifungal agents and ferulic acid. As drug delivery systems, transferosomes showed potential for providing controlled release of drugs and enhancing the stability of unstable compounds. They also made it possible to transport very large molecules, which normally could not diffuse through the skin, across the skin barrier126-129.
Over the past few decades, the use of transferosomes in transdermal drug delivery has been widely explored. Some of these applications are highlighted in the section below.
Transferosomes have proven to be an effective tool for the transdermal delivery of a wide range of therapeutics, including hydrophilic drugs, large molecules, peptides, and proteins. They have been particularly useful as carriers for proteins and peptides, which are large biogenic molecules that are difficult to deliver into the body. When taken orally, these molecules are rapidly degraded in the gastrointestinal tract, which is why they are still primarily administered through injections. Transferosomes help overcome this challenge by enhancing stability and protecting the active compounds from degradation caused by oxidation, light, and temperature. As a result, they offer a promising delivery system for proteins and peptides.
The delivery of insulin through transferosomes has emerged as a successful non-invasive approach for administering large molecular weight drugs via the skin. Traditionally, insulin is given by the subcutaneous route, which is often inconvenient for patients. Encapsulating insulin into transferosomes (known as transfersulin) helps overcome these challenges. Transferosomes improve the bioavailability of insulin by protecting it from degradation in the gastrointestinal tract and enhancing its absorption through the intestinal mucosa. They also help reduce side effects, such as hypoglycemia, by regulating the rate of insulin absorption and maintaining stable blood glucose levels. Furthermore, transferosomes make oral delivery of insulin possible, offering a patient-friendly and less invasive alternative to injections.
Transferosomes have been used to deliver interferons, such as leukocyte-derived interferon (INF), a natural protein with antiviral, antiproliferative, and immunomodulatory effects. Their application in transdermal drug delivery systems (TDDS) shows great potential, as they not only enable controlled drug release but also enhance the stability of sensitive medications.
Transferosomes are a type of vesicular system that have been widely studied for delivering drugs such as corticosteroids through the skin. They enhance site-specific delivery and improve the safety of corticosteroid therapy by optimizing the dose applied on the skin surface. This makes them a promising strategy for transdermal drug delivery, as they help overcome the skin’s natural barrier against large molecules and hydrophilic drugs. Studies have demonstrated that transferosomes not only improve site-specific delivery but also increase the permeation and therapeutic effectiveness of corticosteroids.
One of the most important applications of transferosomes is in transdermal immunization, where they are used to deliver soluble proteins such as integral membrane proteins, human serum albumin, and gap junction proteins. This approach offers two key advantages: it eliminates the need for injections and can produce relatively high antibody titers, including elevated IgA levels.
Transferosomes, a lipid-based drug delivery system, have been investigated for their potential in delivering anesthetics. Studies have shown that they can significantly enhance the penetration of local anesthetics into the skin, leading to a longer-lasting anesthetic effect compared to conventional creams. This makes transferosomes a promising option for topical anesthetic delivery, with the potential to improve both efficacy and duration of action. Remarkably, transferosomal formulations can provide topical anesthesia in less than 10 minutes, achieving about 80% pain insensitivity, comparable to a subcutaneous bolus dose. Moreover, they offer a prolonged duration of action, making them highly advantageous for clinical use.
Many non-steroidal anti-inflammatory drugs (NSAIDs) are associated with significant gastrointestinal side effects. Transdermal delivery using transferosomes offers a promising way to overcome these challenges. NSAIDs are widely used for managing inflammation, pain, and fever, and they have also been investigated for their potential role in treating conditions such as neurodegenerative disorders, cardiovascular diseases, diabetes, and cancer.
Transferosome technology has been explored for the transdermal delivery of anticancer drugs such as methotrexate, showing encouraging results and presenting a promising therapeutic option, particularly for skin cancer treatment. Studies have also demonstrated the potential of transferosomes in delivering plant-derived compounds with anticancer properties, along with other therapeutic agents. These findings suggest that transferosomes could serve as an effective system for the targeted delivery of anticancer drugs, highlighting the need for further research in cancer therapy.
Transferosomes, a type of vesicular carrier, have gained attention for their use in delivering herbal medicines through the skin. Along with other vesicular systems such as phytosomes, niosomes, ethosomes, and cubosomes, transferosomes have been shown to enhance the transport of phytoconstituents across the skin, improving their bioavailability and therapeutic effectiveness. This strategy helps address the common challenges of orally administered herbal extracts and phytoconstituents, including poor lipid solubility, large molecular size, and first-pass metabolism, ultimately leading to better patient compliance and improved therapeutic outcomes.
Transferosomes loaded with an antifungal agent were prepared using the Rotary Flask Evaporation–Sonication method. To optimize the formulation, the Plackett-Burman design was applied to identify key formulation and process parameters influencing vesicle size, including the lipid and surfactant content, ethanol and hydration medium volume, and hydration time.
In a comparative study, ferulic acid an antioxidant derived from natural sources was incorporated into transferosomes and monoolein aqueous dispersions (MADs). The results showed that transferosomes containing poloxamer 188 formed a multilamellar system, which effectively controlled drug release. The choice of non-ionic surfactant was found to influence the release rate, while patch tests confirmed that all transferosome formulations were safe when applied under occlusive conditions for 48 hours.
In a study, carvedilol—a drug with low oral bioavailability (25–35%) was encapsulated in nanostructured lipid carrier (NLC)-loaded transferosomes using a Box–Behnken design. The optimized formulation demonstrated enhanced dermato-pharmacokinetic and pharmacodynamic performance compared to the conventional formulation.
In another study, transferosomes were optimized for the delivery of RotigotineHCl and Rasagilinemesylate. The Plackett–Burman and Box–Behnken designs were employed for formulation screening and optimization. The optimized transferosomes were found to be spherical with a uniform size distribution, making them suitable for drug delivery. Evaluations of vesicle size, entrapment efficiency, and in vitro drug permeation demonstrated promising results for the effective delivery of these drugs.
The ability of transferosomes to target peripheral subcutaneous tissues is attributed to their minimal drug clearance by the blood vessels within the subcutaneous layer.
Transferosomes have been extensively investigated for transdermal delivery because of their remarkable ability to penetrate deep into the skin. For example, diclofenac-loaded transferosomes have shown effectiveness in pain management.
Transferosomes act as effective carriers for antigens, making transdermal vaccination possible and capable of inducing strong immune responses. For instance, influenza and hepatitis vaccines delivered through transferosomes have shown promising results.
Transferosomes enhance drug delivery to the cornea and intraocular tissues, helping to overcome the limitations of conventional eye drops. This approach improves ocular drug bioavailability and therapeutic effectiveness. For example, timolol-loaded transferosomes have been successfully used in the management of glaucoma.
Transferosomes are gaining attention in the cosmetic industry for delivering anti-aging agents and essential nutrients deep into the skin. For instance, vitamin C-loaded transferosomes have demonstrated improved skin penetration along with enhanced antioxidative effects.
Transferosomes have transformed transdermal drug delivery by making it possible to transport large molecules such as insulin and hormones. They are particularly useful for drugs with poor bioavailability. For example, insulin-loaded transferosomes have shown potential for oral diabetes therapy156.
A wide range of drugs have been successfully loaded into transferosomes, allowing for targeted and controlled delivery as shown in following table157,158.
|
Sr. No |
Drug |
Category |
Results |
|
1 |
Repaglinide |
Anti-hypoglycemia drug |
Improved site specificity and prolonged the release of the drug |
|
2 |
Lidocaine |
Local anesthetic |
Improved skin permeation |
|
3 |
Itraconazole |
Antifungal drug |
Prolonged release of the drug |
|
4 |
Carvedilol |
β- Blocker |
Transferosomal vesicles were substantially more effective at delivering carvedilol through the nose with a bioavailability of 63.4% |
|
5 |
Insulin |
Anti-diabetic |
prolonged hypoglycemic effect in diabetic rats over 24 h after transdermal administration |
|
6 |
Mefenamic acid |
NSAID |
Better outcomes were obtained using the thin-film hydration technique, which had the maximum drug content, spread ability, and sustained drug release profile for 12 hours. |
|
7 |
Sildenafil citrate |
PhosphoDiesterase (PDE) Inhibitors |
Improved transdermal permeation and bioavailability with reduced dose administration frequency |
|
8 |
Tacrolimus |
Immunosuppressants |
Better antipsoriatic activities compared to liposomes due to better skin permeations |
REFERENCES







Chaithra R. P., Hafsa H., G. K. Sarpabhushana, Dhananjaya M., Bhoomika K. M., Prajvith V., Kishor Rahul R. P., Review on Transferosomes: Ultra-Deformable Vesicles for Enhanced Transdermal Drug Delivery, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 10, 2039-2076. https://doi.org/10.5281/zenodo.17386337
 10.5281/zenodo.17386337
10.5281/zenodo.17386337