
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1K.B. Institute of Pharmaceutical Education and Research.
2Manager pharmacovigilance, Neocubes Pharma LLP
3Co-CEO-Neocubes Pharma LLP. Ahmedabad
4Assistant Professor, K.B. Institute of Pharmaceutical Education and Research
This abstract explores the integral role of Periodic safety update reporting in the post-marketing surveillance process, highlights the challenges faced by marketing authorization holders in compiling and submitting these reports to Repository, and examines the harmonization of requirements across Europe Union. Pharmacovigilance has grown in breadth throughout time as a result of the realisation that a systematic strategy is necessary to monitor and improve the safe use of medications. Aggregate reporting plays a crucial role in assessing the benefit/risk ration of a medicinal product throughout its lifespan. It is frequently not enough for marketing authorisation holders to thoroughly evaluate the benefit/risk profile and obtain a complete understanding of the safety features of a pharmaceutical product. Therefore, periodic examination and analysis of cumulative safety data from several sources is required. The results are subsequently sent to regulatory bodies in the form of aggregate reports. Periodic Safety Update Reports playing a critical role in the ongoing assessment of medicinal products after they have been authorized for use. These reports are designed to provide regulatory authorities with a comprehensive, cumulative review of safety data, including adverse drug reactions, emerging safety signals, and evolving risk factors. This report contributes significantly to ensuring patient safety and public health by enabling timely interventions, such as label modifications, risk minimization strategies, or even market withdrawal, when necessary. This study emphasizes the importance of continuous safety monitoring and the adaptation of regulatory processes to keep pace with emerging safety data.
The first step in pharmacovigilance activities is Getting safety information from a range of sources, including published journal papers, unplanned reports, regulatory bodies, hospitals, and clinical trials. Each of these reports needs to be ranked and verified for a causality relationship to the product before being added to the pharmaceutical safety database. Following that, each safety report is stored in a drug database. Furthermore, each of these data—either alone or as part of aggregate reports must be provided to the appropriate regulatory agencies and interested parties during the allotted time periods.
There are two kinds of aggregate reports across globe:
The Pharmaceutical product is only approved if the advantages outweigh the hazards. This equilibrium is accurate at a certain time gap but may alter once the product is in the market owing to a high number of patients, concurrent drugs and disorders, long-term exposure, confounding variables, and uncontrolled situations. As a result, several drugs that initially showed great success in many patients were later discovered to have severe side effects, leading to their removal from the market. Therefore, it is crucial to routinely reevaluate the balance to make sure it stays favourable in order to improve patient health. The Periodic re-examination is given in a file called a “periodic safety update report”/PADER/PBRER, during the post authorization phase2.

Periodic Safety Update Report:
Prior to 2012, PSUR filings followed a standardized schedule: every 6 months for the first 2 years of market knowledge, then annually for the next 2 years, and finally every 3 years3. Because of the EU Pharmacovigilance policy, the EMA created and published the first version of the EURD list in 2012. The frequency of PSUR submissions for authorised active ingredients and active ingredient combinations across the EU was included in this list. The EURD list was developed with the intention of standardising data lock points (DLPs, the deadline for records to be entailed in a PSUR) and the intervals of PSUR submissions for the same active ingredients and combination of them in order to facilitate a single EU assessment of the risk-benefit equilibrium of an active ingredient based on all available inputs. MAH are required to deliver relevant PSURs in accordance with the guidelines outlined in the "EURD list" for products that contain active ingredient and combinations of them4. The list is sorted in alphabetical manner. The risk-based methodology outlined in the Guidelines determines how frequently PSURs must be submitted, and considers the following prioritization criteria:
“Any change in these criteria for a given substance may lead to an amendment of the respective entries in the EURD list (e.g., increase or decrease of the frequency for PSUR submissions)5” While other MAHs have authorised an active ingredient in multiple Member States, MAH "X" only has a medicinally approved product in one Member State. Additionally, MAH "X" ought to adhere to the EU PSUR single evaluation procedure and the EURD list.
MATERIALS AND METHODS
The EURD list gives information on following:
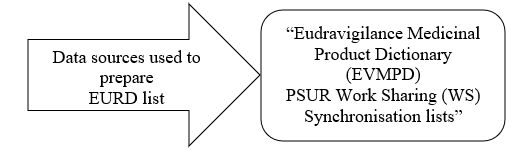
The list is updated monthly by EMA. You can also download the latest list through the https://www.ema.europa.eu/en/documents/other/list-european-union-reference-dates-eurd-frequency-submission-periodic-safety-update-reports-psurs_en.xlsx.
Information significant for the submission of Periodic safety update report
If a fixed pair ingredients are not present in the EURD list, the MAH should notify the agency. PSURs should not be deliver using the EURD list item for one or more individual components of the fixed dosage combination.
To streamline the submission process, each entry and procedure in the EURD list is given a unique procedure number, structure is as follows: “PSUSA/00000000/YYYYMM”, where “PSUSA” denotes a single PSUR assessment procedure, the eight-digit number represents a unique identifier for a specific entry in the EURD List, and "YYYYMM" corresponds to the year and month of the Data Lock Point as published in the EURD list. The MAH should notify the Agency if a certain fixed combination medication is not Present in the EURD list16.
Principles for the preparation of PSURs (VII.B.3.)
The marketing authorization holder is required to prepare a single PSUR for all medications that contain the same active ingredient, unless instructed differently by the appropriate authorities. Regardless of whether the products are approved under various names or through different processes, this report must include all approved indications, dosage forms, methods of administration, and dosing schedules. If applicable, information unique to a certain indication, dosage form, administration method, or dosage schedule needs to be included in a new PSUR heading, with safety issues handled appropriately. Separate PSUR preparation might be deemed appropriate in certain circumstances, such as when there are several formulations for completely different indications. In certain situations, prior consent from the appropriate authorities should be sought, preferably at the time of authorization. The reference product details should include a comprehensive list of all authorized applications across ICH countries or regions. Depending on the preference of the MAH, if the PSUR is also submitted to other nations where additional indications have been approved locally, these may be included in the reference product detail or supplied separately as a regional appendix. Any substantial revisions made during the submission period must be clearly outlined in PSUR Section 4 ("Changes to the Reference Safety Information") and, if relevant, further addressed in PSUR Section 16 ("Signal and Risk Evaluation”) Such revisions may encompass:
A clean copy of every version of the “reference product information” that was in force at the conclusion of the reporting timeframe (for example, for various dosage forms included in the same “PSUR”) must be supplied by the MAH as an appendix to the PSUR. A PSUR created for a generic product should adhere to the format and content defined in GVP Module VII5.
Repository
All MAHs operating within the European Union (EU) are required to submit PSURs for human pharmaceuticals directly to the “PSUR repository” as of June 13, 2016. This implies that businesses should no longer send PSURs to NCAs directly and instead use the repository as a central location for all submissions. Direct, secure access to the repository is available to National Competent Authorities (NCAs). All users should register using the self-registration feature ahead of they may submit a PSUR to the PSUR repository either the eSubmission Gateway or Web Client. PSURs have to be submitted as either a non-eCTD electronic submission (NeeS) or an Electronic Common Technical Document (eCTD). Submissions of PSURs in any other format will be refused and cannot be published to the PSUR repository.
eCTD Format Includes:
The Repository screen is divided into five sections:
Flowchart of submission:
The submission of PSUR is not only publishing the eCTD sequence!
Cover letter- format given by EMA
PSUR document- Prepared by MAH using PV databases (e.g. Argus/ Pvedge)
Using knowledge net software/ any other software
RESULT AND DISCUSSION
eCTD Sequence publishing:
In any eCTD publishing PSUR is never been submitted in initial submission because it is a part of post authorization phase. That is why we have to freeze the old sequence and then publish it as a fresh for next sequence. For freezing the old sequence, you have to click on submission in that click on publish and submit, find your project in it and click on confirm to freeze it.
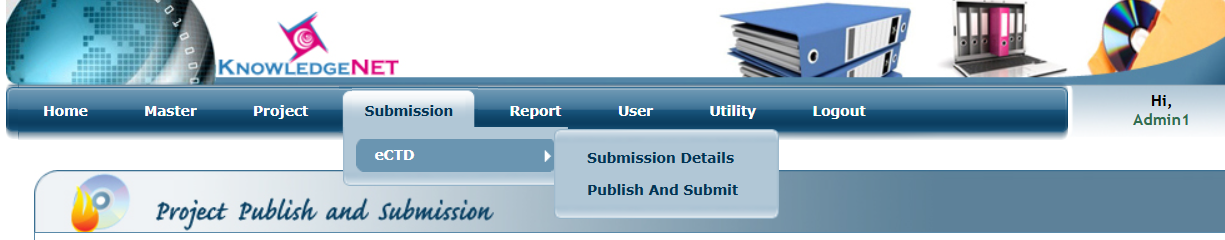
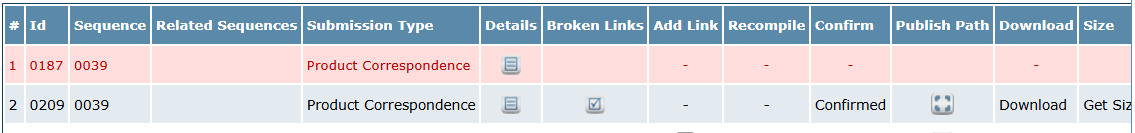
Figure 1: Publish and submit interphase in eCTD software
Add cover letter in m1 select specific country and repeat the node, add PSUR document in Additional data section if country or procedure specific otherwise add it to m536 with appropriate naming, The naming of the leaf element in m536 shall indicate the number of the PSUR or the period covered. After document uploading clear your work from my pending work. Publish the sequence by adding submission and publishing details.
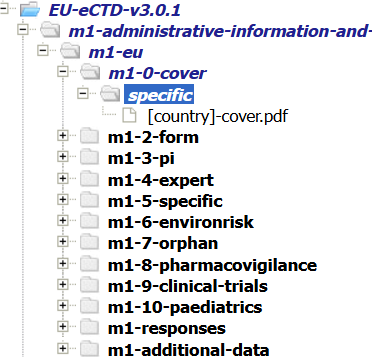
Figure 2: EU Module 1 in eCTD software
For CAPs, the proposed product information should also be submitted to Module 1.3.1 of the eCTD. It should be presented as a tracked change version of each EU SmPCs and package leaflet of the products concerned and each product information should be translated into English language including the tracked changes proposed, in order to enable the EU single assessment. This can result in having to submit a large number of sets of tracked change product information with the additional burden of providing translations. Hence MAHs can consider the option to focus on the proposed amendments to SmPC and package leaflet. In such case, only the amended parts of the SmPC and package leaflet should be provided in track changes and in English language under the EU regional appendix. Where the proposed changes are not based on the data submitted within the PSUR, these will not be considered, and a variation will have to be submitted as appropriate to the relevant national competent authority.
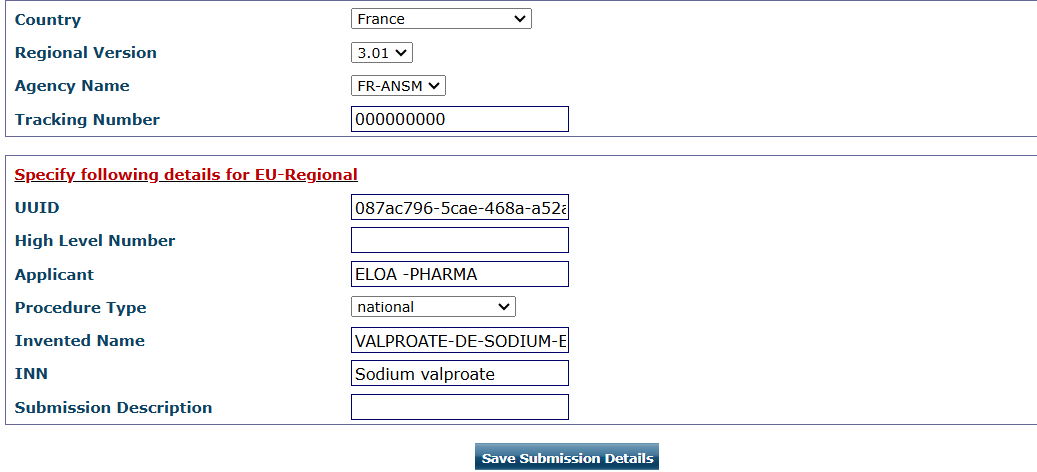
Figure 3: Submission detail interphase in eCTD software

Figure 4: Publish and Submit interphase in eCTD software
Repository Section
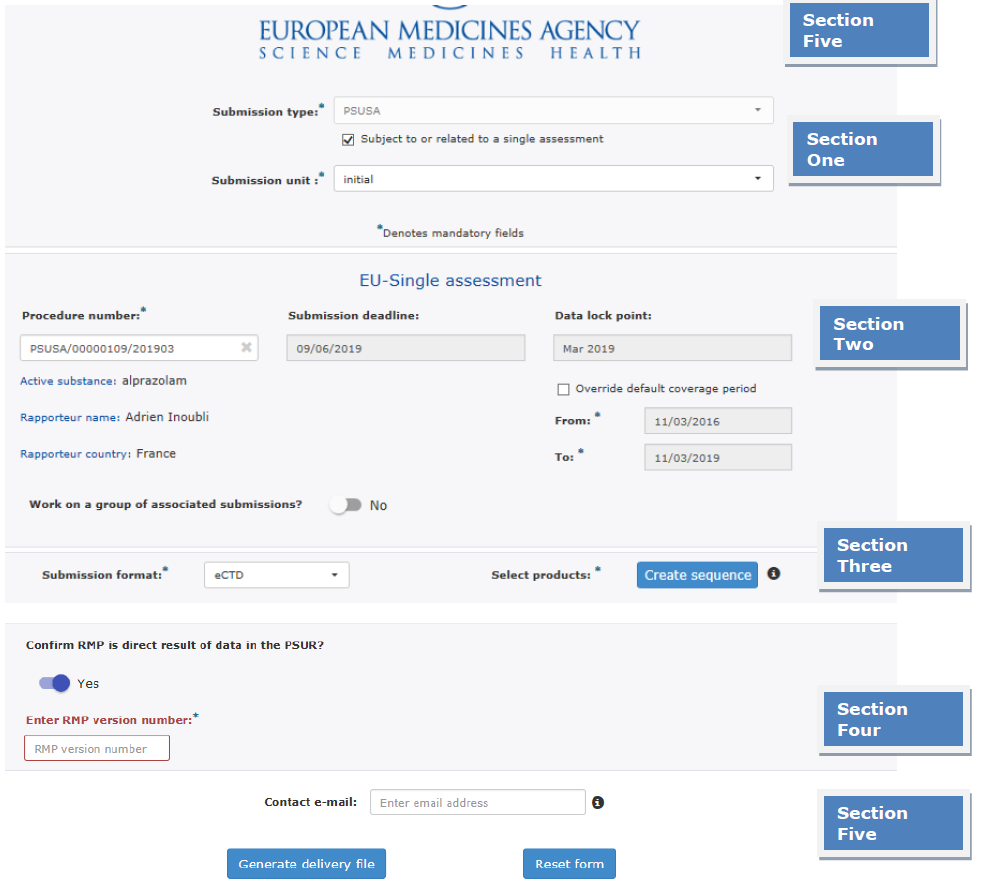
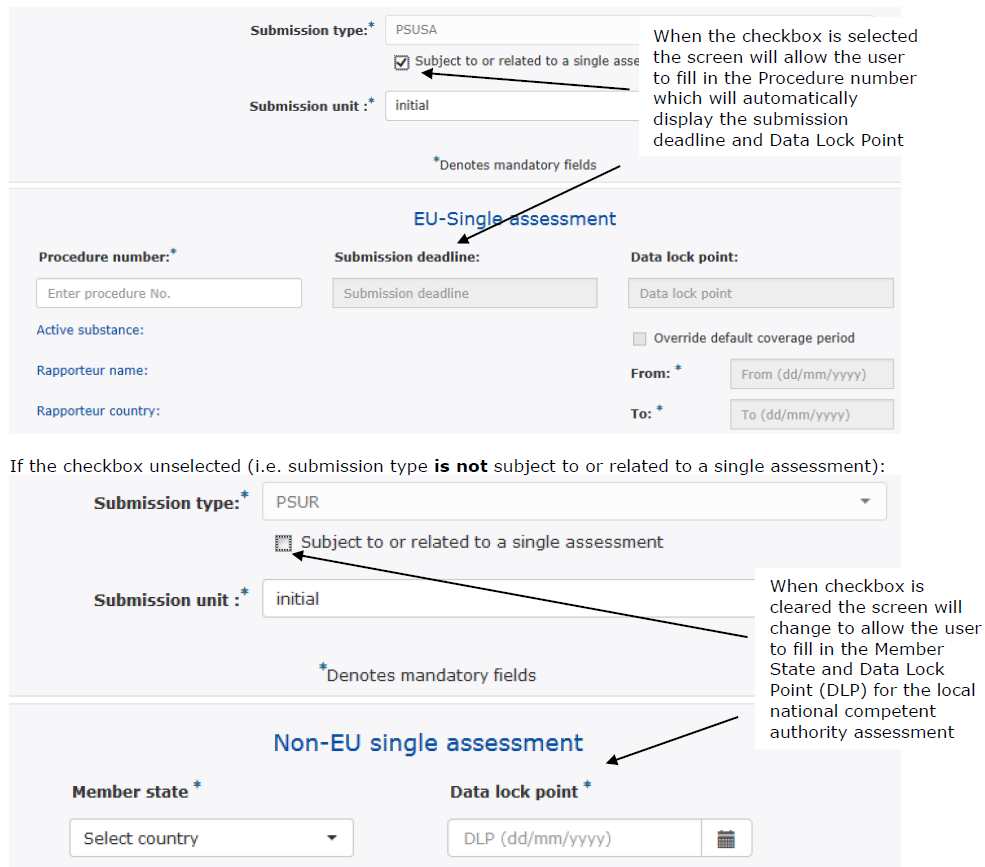
Figure 5: Repository interphase
If you have missed the submission deadline, please always request a Late Submission ID via the EMA Service Desk. Late submissions must not be submitted using submission unit ‘response’.
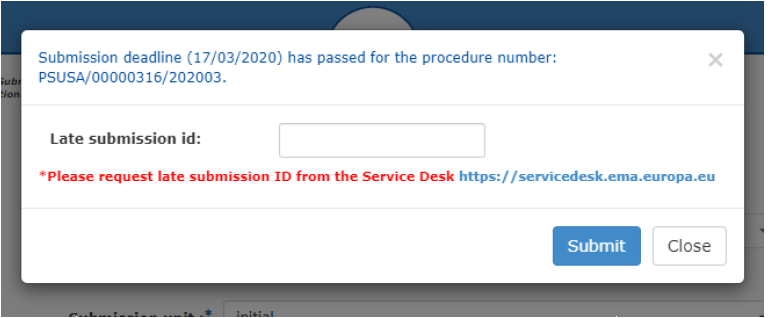
Figure 6: Late submission ID phase
If you want to group together associated submissions i.e. you have created just one single PSUR covering multiple products that have different eCTD sequences.


Figure 7: Group working interphase
Generate a ‘New Group ID’ or use one you have previously generated and noted down if one of the submissions for associated products has failed.
Figure 8: Submission format
Click ‘Create sequence’ to display the list of products containing the relevant active substance(s) based on the procedure number from the EURD list.
Figure 9: Sequence Creation
The system will display a list of products that are included in the procedure and that containing the relevant active substance(s). The products are presented with all presentations, strengths and formulations.
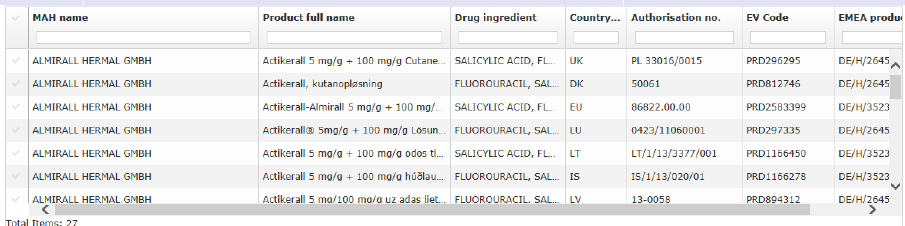

Figure 10: Product selection
Only one sequence number can be entered per product selection window.
Figure 11: Contact e-mail
Enter email address of the person who is the responsible contact for the PSUSA procedure.All relevant correspondence will be sent to this email address. Click “Generate Delivery File” and save the file to your local machine.
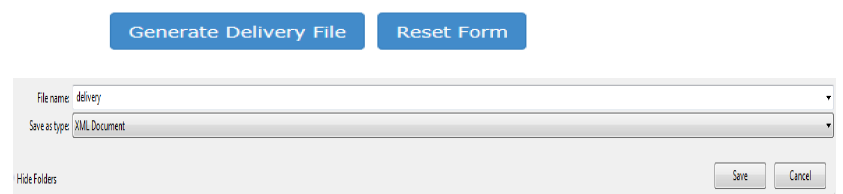
Figure 12: Create delivery file screen
Add the delivery file to the submission ZIP file package on the root level. For eCTD submissions for CAPs and NAPs that do not have harmonised lifecycle, only one product (with all its presentations) can be selected.
The process of Preparation to Submission of periodic safety update reports is completed. After successful submission the applicant will receive an acknowledgement through Repository.
CONCLUSION
The project highlights the importance of PSURs in the ongoing assessment of drug safety and regulatory compliance. It underscores both the impact of effective PSUR reporting on public health and regulatory decisions, as well as the significant challenges in ensuring accurate, timely, and globally compliant reporting to Repository. Addressing these challenges is essential for improving the drug safety monitoring process and ultimately protecting patient well-being. PSURs help authorities make informed decisions about the ongoing safety of drugs. The impact on regulatory actions, such as approvals, suspensions, or additional warnings, is a central focus. PSURs require considerable effort and resources to compile. This includes collecting adverse event data, interpreting results, and ensuring the safety of marketed products. Additionally, there are complexities in managing the volume of safety data, which can lead to delays and inefficiencies in reporting. Future advancements will likely focus on streamlining processes, incorporating real-time data, and ensuring that safety reporting aligns more seamlessly with global standards, ultimately enhancing drug safety surveillance and patient protection.
REFERENCES


Sakshi Joshi*, Arnab Majumedar, Bhavik Joshi, Vinit Movaliya, Impact and Challenges of Periodic Safety Update Reporting, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 3, 2717-2726. https://doi.org/10.5281/zenodo.15098812
 10.5281/zenodo.15098812
10.5281/zenodo.15098812