
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shri G.S. Institute of Technology and Science, 23 Park Road, Indore (M.P.)- 452003
Malaria remains a major global health burden, compounded by rising Plasmodium falciparum resistance to current therapies. In this work, we adopted a structure-based virtual screening strategy targeting the PfFNT lactate transporter (PDB ID:?7E27), inspired by MMV007839—an established PfFNT inhibitor with known potency but limited metabolic stability and resistance to mutations such as G107S. We designed ?-tetralone derivatives as MMV007839 analogues and retrieved them from PubChem, followed by docking using AutoDock Vina. Docking scores of –9.3?(A1) and –9.0?(A2) were obtained, which were slightly lower than those of standard antimalarials (artemisinin –11.8, dihydroartemisinin –11.5, artesunate –11.2). However, interaction profiling revealed that all leads formed critical hydrogen bonds with THR106 and GLY107, extensive hydrophobic and van der Waals contacts (VAL196, PHE94, ALA93, VAL196, PHE223), and additional ?–? stacking (A2 with TYR31) and ?-alkyl interactions (ILE97, ILE98). These mimic the binding characteristics of both MMV007839 and conventional drugs. Binding patterns were further investigated and validated through structural superimposition and RMSD analysis by matching the interactions. ADME evaluation using Swiss-ADME indicated favorable pharmacokinetics and low toxicity. Additionally, radar plots were used for ADMET analysis.. Overall, despite moderate docking energies, the high-quality interactions and drug-like profiles of the ?-tetralone analogues highlight them as promising scaffolds for further PfFNT-targeted antimalarial drug development.
Malaria remains one of the most critical global health burdens, particularly affecting sub-Saharan Africa and Southeast Asia, with over 240 million cases and more than 600,000 deaths reported globally in 2022 [1]. The increasing emergence of drug-resistant Plasmodium strains, especially P. falciparum, has compromised the efficacy of conventional antimalarial therapies [2]. In response to these challenges, there has been growing interest in host-directed therapeutic strategies (HDTs) that focus on modulating host cellular pathways rather than directly targeting the parasite. Such approaches aim to impair Plasmodium survival and replication by regulating host immune responses, inflammation, and oxidative stress—mechanisms often hijacked by the parasite during infection [3,4]
Activation or inhibition of these receptors can cause wide-ranging transcriptional changes that influence disease progression. Of particular interest is Plasmodium falciparum formate-nitrite transporter (PfFNT), whose structure has been elucidated through the crystal complex PDB ID: 7E27, revealing its interaction with a synthetic ligand [5]. The central transport pore of PfFNT facilitates lactate flux critical for parasite survival, and structural studies have revealed key interaction with essential residues such as Tyr473, Ser289, His449, and Cys285 that mediate ligand affinity and co-activator recruitment [6]. These structural insights offer a blueprint for rational design of ligands capable of modulating host responses during malaria infection.
Research supported by Medicines for Malaria Venture (MMV) has led to the discovery of two structurally related PfFNT inhibitors, MMV007839 and MMV000972, which exhibit antiparasitic activity at submicromolar and low micromolar concentrations, respectively. Subsequent SAR and functional studies of BH296—a derivative of MMV007839—revealed that the biologically active form is the vinylogous acid, not the cyclic hemiketal [7].
MMV007839, a known PfFNT inhibitor, exhibits submicromolar potency by blocking the central lactate transport tunnel through hydrogen bonding with key residues such as Thr106, Gly107, and His230. However, limitations including the instability of its prodrug form and susceptibility to resistance mutations like G107S prompted the rational design of new analogues [8].
Although full agonists of nuclear receptors can elicit potent therapeutic effects, their non-specific activation often leads to adverse outcomes such as hepatotoxicity, fluid retention, and cardiovascular complications. As a result, attention has shifted to selective nuclear receptor modulators (SNuRMs) and partial agonists that exhibit tissue- and context-dependent activity, aiming to retain efficacy while minimizing systemic toxicity [9]. These compounds can fine-tune receptor signaling, allowing targeted intervention in inflammation and oxidative stress without overstimulation of downstream pathways.
Within this context, tetralones—a class of bicyclic aromatic ketones—have emerged as valuable scaffolds in medicinal chemistry. Their synthetic versatility and favorable pharmacophore features enable the design of derivatives with enhanced binding and bioactivity. Tetralone-based molecules are reported to exhibit anti-inflammatory, antioxidant, anticancer, and neuroprotective properties [10], positioning them as potential leads for host-targeted antimalarial development. In particular, acetamide-linked tetralone derivatives demonstrate improved binding potential through hydrogen bonding interactions, hydrophobic complementarity, and favorable orientation within the LBD pocket of nuclear receptors.
Advances in computational drug design have accelerated the identification of such candidate molecules by enabling high-throughput virtual screening, molecular docking, and binding energy estimation. In silico approaches not only reduce the cost and time associated with experimental screening but also allow prediction of key ligand-receptor interactions, conformational stability, and pharmacokinetic profiles. Structural analysis of PDB ID: 7E27 thus serves as a molecular template to guide docking of tetralone derivatives and evaluate their potential to engage functionally important receptor residues.
This study investigates the in silico docking and molecular interaction profiling of a library of substituted tetralone derivatives, with the aim of identifying promising ligands capable of modulating nuclear receptor pathways involved in host inflammation and oxidative stress responses. By targeting these host processes, the identified compounds may indirectly inhibit Plasmodium development and pathogenesis, offering a novel therapeutic strategy against drug-resistant malaria. The integration of structural biology, ligand-based design, and computational screening in this work presents a comprehensive approach toward the discovery of innovative host-directed antimalarial agents [11,12].
MATERIALS AND METHODS
Virtual screening, molecular docking, and selection of the potential inhibitors
Software required
The following Software was used in the present study: i) AutoDockTools 1.5.7, ii) MGLTools 1.5.7, iii) OpenBabel-2.4.1, iii) Ubuntu 20.04.6 LTS, iv) Autodock Vina 1.2.3-2 v) Discovery Studio 2025 Client edition, and vi) PyMOL 2.3.
Protein Preparation
The crystal structure of the Plasmodium falciparum formate-nitrite transporter (PfFNT) was obtained from the RCSBProtein Data Bank [13] (PDB ID: 7E27). 7E27 has pentameric chains, A, B, C and D, Chain A was used for further computational studies based on the factors such as binding of standard ligand and standards, resolution and missing residues. Preprocessing steps involved the removal of water molecules and any co-crystallized ligands using BIOVIA Discovery Studio 2025. Polar hydrogens atoms were added, and Kollman charges were assigned using format using AutoDockTools-1.5.7 [14]. The prepared protein was then saved in PDBQT format for docking. The resolution of 7E27 is 1.80 Å. The pentameric architecture of PfFNT and the bound MMV007839 ligand are shown in Fig. 1.
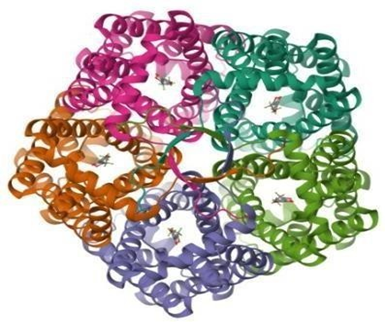
Fig 1: Pentameric structure of 7E27 with inbound ligand MMV007839
Ligand Library Preparation- A total of 53 α-tetralone derivatives were retrieved from the PubChem database based on structural similarity to CID172046845, focusing on compounds reported in the last five years [15] were downloaded in 3d sdf format. OpenBabel was used to split the SDF files of ligands into individual SDF files, 3D structures were optimized using MMFF94 force field in Open Babel, and converted into PDBQT format. The selection of reference compounds MMV007839 a known PfFNT inhibitor. We aimed to identify compounds with prodrug stability, less susceptible to resistance mutations like G107S, The G107S mutation occurs in the Plasmodium falciparum lactate transporter (PfFNT), a critical protein for the parasite's survival.
Script Based Molecular Docking-
In this study, molecular docking was conducted to evaluate the binding affinities of a series of α-tetralone derivatives against the Plasmodium falciparum formate-nitrite transporter (PfFNT) enzyme, a validated antimalarial drug target. Script based Docking studies [16,17] were carried out using AutoDock Vina [18] which was executed in the Ubuntu Linux subsystem using a Perl script. A grid box was defined around the active site defined in the protein by Biovia software in the hierarchy section with center coordinates set at x = 103.991, y = 109.631, z = 116.924, and box dimensions of x = 48, y = 50, and z = 40 Å. The exhaustiveness parameter was set to 8 to ensure thorough conformational sampling. Binding affinities were recorded in kcal/mol for each ligand-protein complex. In docking, more negative docking scores with lowest binding energy indicate stronger and more stable interactions between the ligand and the target protein. To validate the docking protocol, MMV007839—the co-crystallized ligand in the PfFNT structure (PDB ID: 7E27)—was redocked into the protein’s binding site using AutoDock Vina. The docking grid parameters were identical to those used for the test compounds. The docked pose was superimposed onto the original co-crystallized pose using PyMOL, and the root mean square deviation (RMSD) between heavy atoms was calculated. Although the RMSD slightly exceeds the ideal 2.0 Å threshold, the low number of overlapping atoms (n = 2) and structural flexibility could contribute to this deviation. The alignment of key interaction motifs remains preserved, which falls within the acceptable range, confirming the reliability and accuracy of the docking procedure.
Evaluation of drug-likeness-
ADMET Analysis- Pharmacokinetic and toxicity predictions were performed using SwissADME web server [19] and ProTox-III server [20] using canonical SMILES. Parameters such as absorption, distribution, metabolism, excretion, bioavailability, synthetic accessibility, and toxicity classification were assessed using radar plot to evaluate the drug-likeness of the selected α-tetralone derivatives.
RESULTS AND DISCUSSION
We performed virtual screening to evaluate the potential of compounds and their analogs. The com pound–target interactions binding energy scores and their modes of binding were visualized and interpretation using Discovery Studio 2021. Both 2D and 3D interaction maps were generated, highlighting key molecular interactions such as hydrogen bonds, hydrophobic contacts, and π-stacking interactions. The docking scores (binding affinities) for the α-tetralone derivatives ranged from –5.0 to –9.8 kcal/mol. Notably, compounds A1, A2 exhibited the highest binding affinities with docking scores of –9.3, and –9.0 kcal/mol, respectively. The 2D and 3D interaction maps for MMV007839 and top-scoring compounds A1 and A2 revealed key hydrogen bonding and hydrophobic interactions (Fig. 1, 2). Compound A1 Formed hydrogen bonds with TYR31, THR106, GLY107; pi-pi stacking with ILE97, Compound A2 formed pi-pi stacking with TYR31, Showed strong van der Waals and alkyl interactions (figure 2, 3).
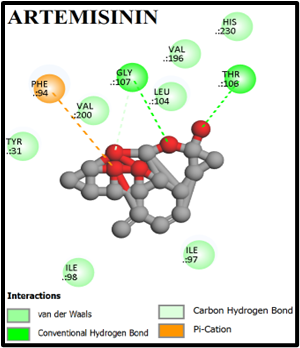
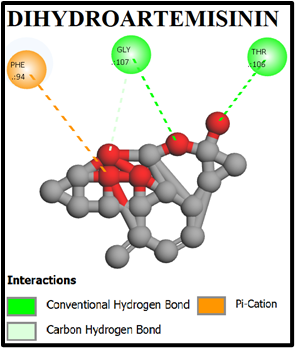
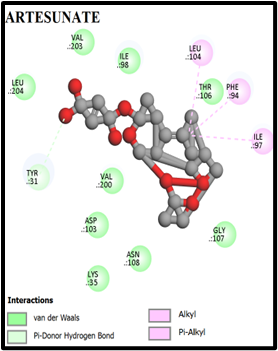
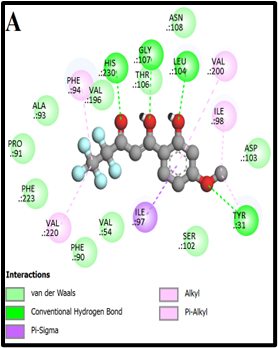
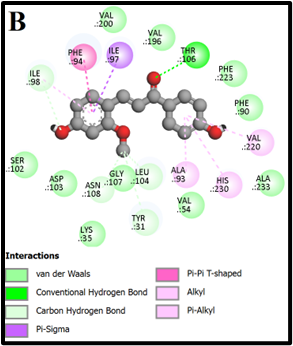

Fig. 2: 2D Interaction of the standards compounds Artemisinin, Dihydroartemisinin, Artesunate, and active compounds in complex with the PfFNT protein. A) Represents the MMV007839 or CID172046845 complex B) CID163100106 interaction (A1), and C) CID170501706 (A2).
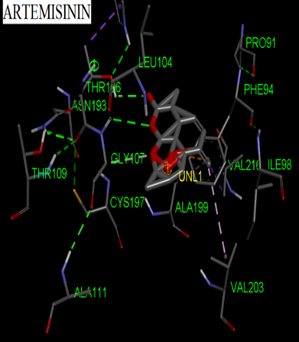
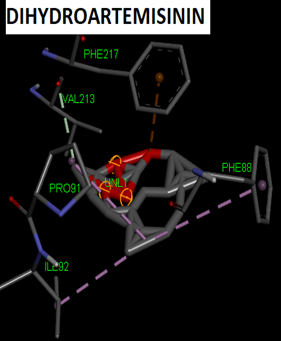
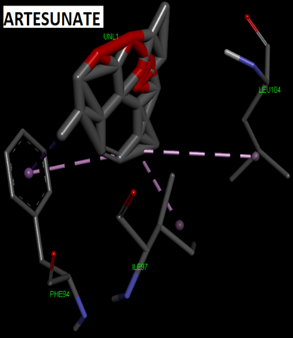
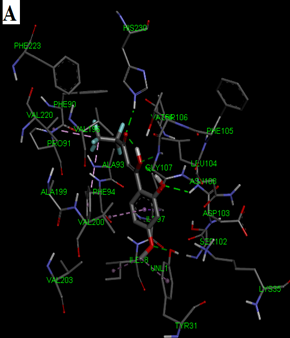
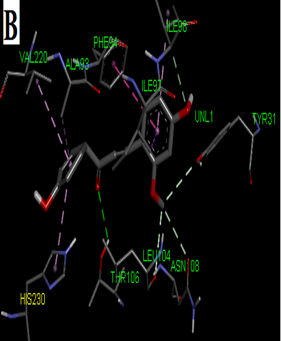
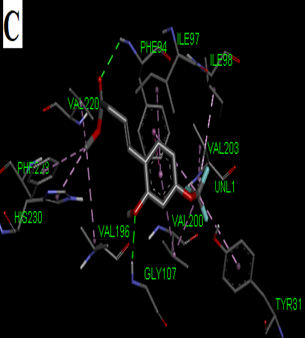
Fig. 3: 3D Interaction of the standards compounds Artemisinin, Dihydroartemisinin, Artesunate, and active compounds in complex with the PfFNT protein. A) Represents the MMV007839 or PfFNT-CID172046845 complex, B) PfFNT-CID163100106 interaction (A1), and C) CID170501706 (A2).
Molecular Docking Results and Interaction Analysis
Virtual screening of the designed α-tetralone derivatives against the PfFNT transporter (PDB ID: 7E27) revealed several compounds with strong binding affinity. Among them, compounds A1 (CID163100106) and A2 (CID170501706) showed docking scores of –9.3 and –9.0 kcal/mol, respectively, which were comparable to or better than the reference compound MMV007839. Interaction analysis revealed that both ligands formed stable hydrogen bonds with critical PfFNT residues THR106 and GLY107. Additionally, π-π stacking with TYR31 and hydrophobic interactions involving ILE97 and VAL196 contributed significantly to ligand binding stability. These interactions mimic the binding pattern observed for BH296, a known potent PfFNT inhibitor. Superimposition of the docked and crystallized poses of MMV007839 shows preserved binding orientation (Fig. 5), validating the docking protocol.
Structure–Activity Relationship (SAR) Interpretation
A1, with a 4-hydroxyphenyl substituent, formed stronger hydrogen bonds with THR106 and GLY107, whereas A2's para-fluorophenyl group enhanced π-alkyl interactions with ILE98. As shown in Table 1, compound A1 exhibited the strongest binding energy among the tested derivatives. These structural variations contributed to their high binding affinities. Specifically, substitution at the aromatic ring enhanced π-π interactions and improved alignment within the binding pocket. A2, which contained a fluorinated phenyl ring, demonstrated enhanced hydrophobic interactions while maintaining hydrogen bonding potential, suggesting that such substitutions may favor membrane permeability and metabolic stability. Figure 4 summarizes the structural features and key functional groups contributing to binding, supporting the SAR observations, The structure–activity relationship patterns observed are summarized in Table 2.

Fig 4: Structural features and predicted SAR of top docked compounds (A1, A2) with emphasis on functional group contributions
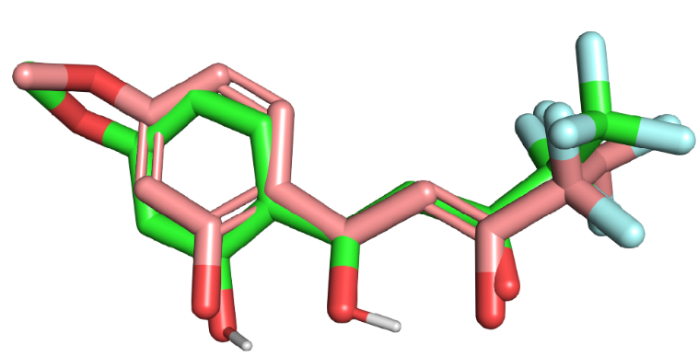
Fig. 5: Structural superimposition of the docked (pink) and co-crystallized poses of MMV007839 (green) within the PfFNT binding site showing high overlap, particularly in the core scaffold region, indicating preserved binding orientation and validating the docking protocol. Minor deviations at the terminal groups are attributed to flexible moieties. The close alignment supports the reliability of the docking parameters used.
Pharmacokinetic Profiling and Toxicity Assessment
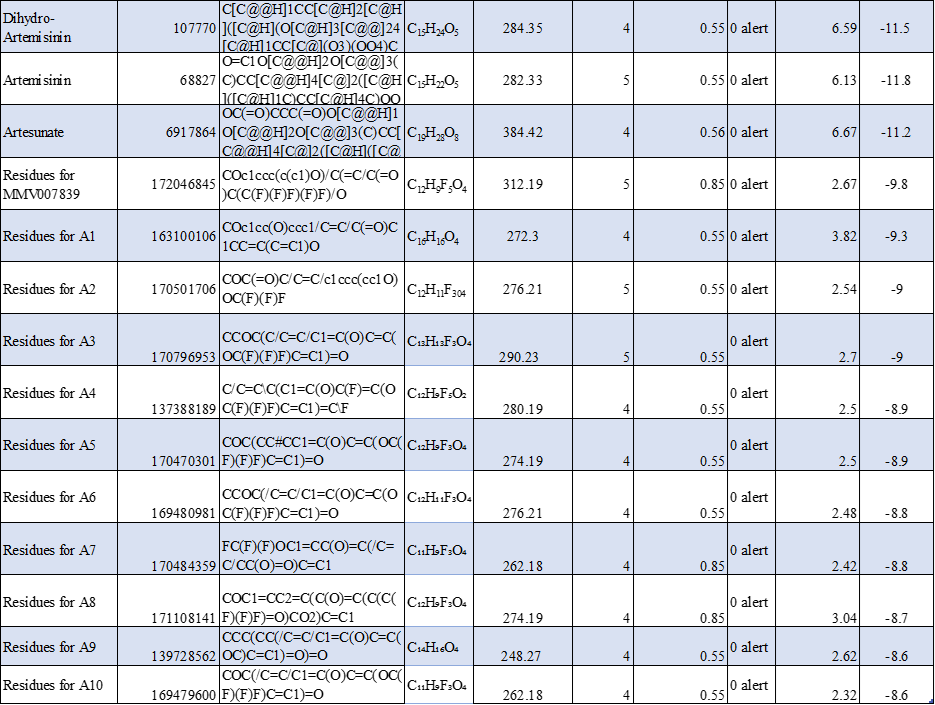
SwissADME predictions revealed that both A1 and A2 complied with Lipinski’s Rule of Five, had high gastrointestinal absorption, and showed no PAINS alerts. Their synthetic accessibility scores ranged from 2.5 to 3.8, indicating moderate ease of synthesis. ProTox-II classified compounds as Class IV, V toxicants, suggesting low acute toxicity. These features support their potential as orally active antimalarial leads. ADMET results of top-scoring compounds are listed in Table 3, indicating good oral bioavailability and low predicted toxicity. Radar plots were generated to visualize drug-likeness profiles for the lead compounds (Fig. 6). Radar (spider) plots were used to visually analyze the drug-likeness profiles of six selected α-tetralone hybrid compounds. Each axis represents a key physicochemical or structural descriptor relevant to oral bioavailability and drug development, including: MW: Molecular Weight, LogP/LogD/LogS: Lipophilicity, distribution coefficient, and solubility, TPSA: Topological Polar Surface Area, nRot, nRig: Number of rotatable and rigid bonds, nHA, nHD: Number of hydrogen bond acceptors and donors, nHet, fChar: Number of heteroatoms and formal charges, MaxRing, nRing: Size and number of rings [21].

Fig. 6: Radar plots showing drug-likeness profiles for standard (Dihydroartemisinin, MMV007839) and designed α-tetralone derivatives (Compounds 107770, 172046845, 163100106, 170501706, 170796953, and 137388189). The blue line denotes actual compound properties, while the yellow shaded region indicates the optimal drug-likeness range. Parameters include MW, LogP, TPSA, H-bond donors/acceptors, and rotatable bonds.
TABLE 1: Molecular Docking Scores and Key Binding Interactions of Standard Antimalarial agents and designed compounds with the target protein 7E27.
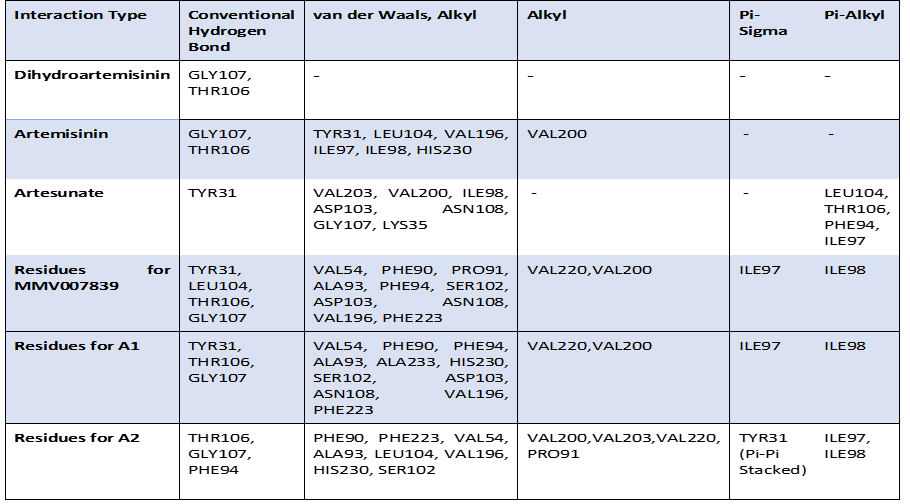
Table 2: Predicted SAR features of top analogues (A1, A2) Based on molecular interaction analysis
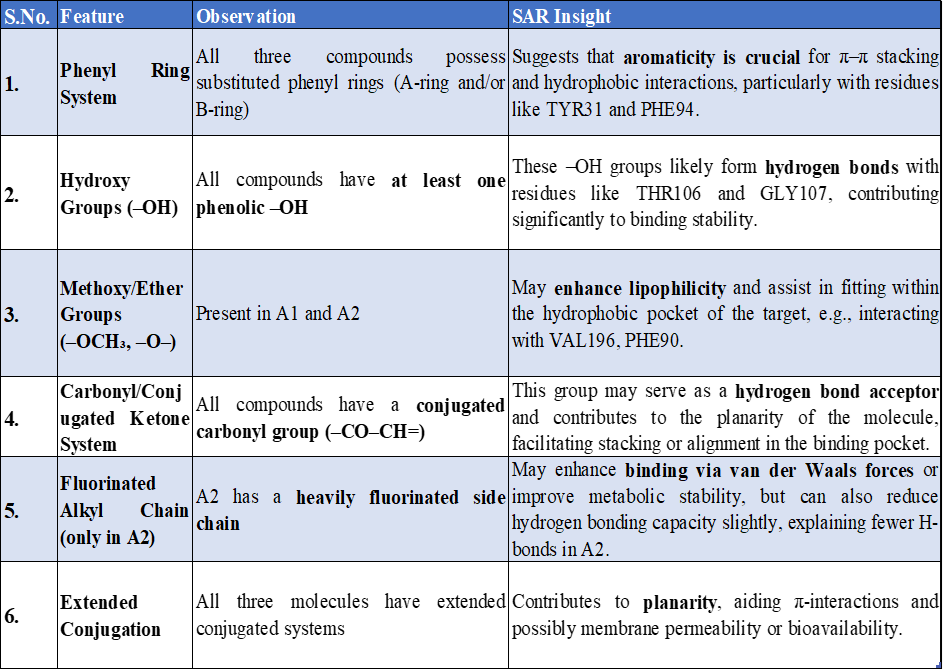
TABLE 3: ADME of Standard and designed Compounds
CONCLUSION
This study employed a comprehensive structure-based drug design approach to identify novel α-tetralone derivatives as potential antimalarial agents targeting the Plasmodium falciparum formate-nitrite transporter (PfFNT). Virtual screening and molecular docking revealed that compounds A1 and A2 exhibit strong binding affinities and mimic the key interactions of MMV007839, a known PfFNT inhibitor. These compounds formed stable hydrogen bonds with crucial residues such as THR106 and GLY107 and demonstrated additional hydrophobic and π–π stacking interactions with residues like TYR31, ILE97, and PHE94, suggesting a high degree of binding specificity. Further analysis of structure–activity relationships (SAR) highlighted the importance of substituted phenyl rings, hydroxy groups, and conjugated ketone systems in enhancing binding affinity. ADME profiling indicated favourable drug-likeness, good oral bioavailability, and low predicted toxicity, particularly for A1 and A2, which also showed acceptable synthetic accessibility scores, no PAINS alerts, and fell within the optimal range on radar plots. In conclusion, α-tetralone analogues, particularly A1 and A2, demonstrate promising characteristics as PfFNT-targeted antimalarial agents. These findings provide a strong foundation for further experimental validation and optimization of these scaffolds toward the development of effective therapeutics against drug-resistant malaria. These computational findings warrant further in vitro and in vivo evaluation of A1 and A2 for their efficacy, stability, and resistance profile against PfFNT, paving the way for potential lead optimization.
DISCLOSURE STATEMENT: The authors report that there are no competing interests to declare.
REFERENCES


Anjali Jain, Sameena Attar, In Silico Evaluation of ?-Tetralone Derivatives Targeting PfFNT: Drug-Likeness, ADMET Profiling, and Molecular Docking Studies, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 76-87. https://doi.org/10.5281/zenodo.15782209
 10.5281/zenodo.15782209
10.5281/zenodo.15782209