1,3Sudhakarrao Naik Institute of Pharmacy, Pusad, Postal address: Nagpur Road, Pusad, Maharashtra, 445204.
2Institute of Chemical Technology, Department of Pharmaceutical Science and Technology, Mumbai.
Itraconazole (ITZ), a poorly water-soluble antifungal drug, exhibits limited oral bioavailability, which hampers its therapeutic efficacy. This study aimed to develop a nanosuspension formulation of ITZ using High Pressure Homogenization (HPH) to enhance its solubility and dissolution rate. Tween 80 was used as a stabilizer, while HPMC E5, Kollidon VA64, and Soluplus served as wetting agents. Process parameters such as stirring time, homogenization speed, and pressure were optimized using a 1:1 drug-to-surfactant ratio. The resulting nanosuspension was adsorbed onto carriers like Neusilin UFL2 and Florite PS200 to obtain free-flowing powder, which was then compressed into tablets. Characterization was performed using particle size analysis, drug content, polarized light microscopy, ATR-FTIR, DSC, and XRD. The optimized formulation showed spherical particles with an average size of approximately 500 nm, good chemical stability, and no significant interaction with excipients. In vitro dissolution studies in 0.1N HCl revealed significantly improved drug release compared to pure ITZ, confirming that nanosuspension via HPH is an effective strategy to enhance the solubility and dissolution profile of poorly soluble drugs like itraconazole.
Oral drug delivery remains the most preferred route for pharmaceutical formulations due to its ease of administration, patient compliance, cost-effectiveness, and flexibility in designing various dosage forms [1-3]. However, the therapeutic performance of orally delivered drugs is frequently hindered by poor water solubility, especially in the case of active pharmaceutical ingredients (APIs) falling under Biopharmaceutics Classification System (BCS) Class II. These drugs are highly permeable but poorly soluble, which restricts their dissolution in gastrointestinal fluids and ultimately results in reduced bioavailability [4-8]. Itraconazole (ITZ) is a broad-spectrum antifungal agent belonging to the triazole class, commonly used in the treatment of superficial, subcutaneous, and systemic fungal infections [9-11]. Itraconazole (ITZ), a BCS Class II drug, possesses a high log P value of 3.7 and exhibits extremely low aqueous solubility (1–4 ng/mL), leading to dissolution-limited absorption and consequently poor and inconsistent oral bioavailability [12-15]. Due to its highly crystalline nature, the solubility and dissolution of itraconazole (ITZ) are significantly limited, making it essential to apply suitable formulation approaches to address these challenges. Similar to other antifungal drugs, ITZ acts by inhibiting fungal cytochrome P450 enzymes, thereby interfering with sterol biosynthesis in the fungal cell membrane and ultimately causing cell death [16-17]. To address the solubility challenges associated with poorly water-insoluble drugs like itraconazole, various formulation approaches such as micronization [18-19], salt formation [20] prodrugs [20], cocrystals [21-22], micelle systems [23], self-emulsifying drug delivery systems [24], particle size reduction (Micronization) [25], drug complexation [26], and solid dispersions [27-28] can be utilized accordingly. Reducing particle size is one of the most established techniques for enhancing drug solubility, as solubility is closely linked to the size of the drug particles. A decrease in particle size leads to an increase in the surface area-to-volume ratio, providing a greater surface area for interaction with the dissolution medium. This enhanced surface area promotes better solvation, ultimately improving solubility. As a result, particle size reduction methods are commonly employed to boost the bioavailability of drugs with poor water solubility [29]. Micronization is a traditional particle size reduction method that is widely employed to enhance the solubility of BCS Class II drugs [30]. For this reason, reducing particle size is considered a reliable and safe approach to enhance the solubility of drug substances without affecting their chemical integrity. It is well established that smaller particle sizes result in a larger surface area, which in turn leads to a faster dissolution rate an observation originally explained by the Noyes–Whitney equation in the late 19th century [31]. While particle size reduction can greatly enhance the dissolution rate, its impact on the solubility of drug substances is relatively limited, as it does not change the solid-state characteristics of the material. However, according to the Ostwald–Freundlich equation, a notable increase in solubility can be observed when the particle size is reduced to the submicron range, particularly below 1 µm (radius < 0.5 µm) [29, 32]. This occurs because reducing the particle size to below 1 μm enhances solvation pressure, which leads to improved solubility, while also disrupting solute–solute interactions, thereby facilitating the solubilization process [33]. Micronization does not alter the drug’s equilibrium solubility but significantly enhances its dissolution rate by increasing the surface area-to-volume ratio, allowing the active pharmaceutical ingredient (API) to dissolve or diffuse more rapidly. Traditional particle size reduction methods in pharmaceuticals involve mechanical processes like milling, grinding, and crushing of bulk materials. These methods reduce particle size through various forces such as pressure, friction, shear, attrition, or impact. Common equipment used for drug micritization includes jet mills, ball mills, and high-pressure homogenizers. Among these, fluid energy milling (jet milling) is the most widely adopted and preferred technique for achieving fine particle sizes in a dry state [34]. Various size reduction techniques have been shown in numerous studies to enhance the dissolution and bioavailability of poorly water-soluble drugs by reducing particle size and increasing surface area. Among these, high-pressure homogenization (HPH) is widely adopted top-down approach is commonly used for the production of nanosuspensions of hydrophobic drugs. This method has been proven effective in significantly improving the dissolution rate and bioavailability of several poorly soluble drugs, including spironolactone, budesonide, and omeprazole, by successfully reducing their particle size into the nano meter range [35]. High-pressure homogenization (HPH) has been recognized for its ability to overcome the limitations of traditional size reduction techniques, which often lead to issues such as amorphization, polymorphic changes, and metal contamination due to the intense mechanical energy involved in conventional milling [36].
Nanosuspension:
This approach is typically applied to drugs that exhibit poor solubility in both water and oil. A nanosuspension is a biphasic delivery system widely used in the pharmaceutical industry, consisting of nano sized drug particles dispersed in an aqueous medium containing surfactants for stabilization. It is suitable for various routes of administration, including topical, oral, parenteral, and pulmonary. The solid drug particles in a nanosuspension generally have an average size between 200 to 600 nm. These systems are developed using either top-down or bottom-up methods. The top-down approach includes several techniques such as nano edge, nano jet, and nano crystal milling [37]. In the current study, an effort was made to develop nanosuspensions of itraconazole using various polymeric carriers at different drug loading levels. These nanosuspensions were subsequently adsorbed onto suitable solid carriers for further processing. Neusilin UFL2 being carrier having a high?surface?area (~400?m²/g), amorphous magnesium aluminometasilicate can adsorb 240–340% of its weight in oil or water while maintaining flow ability and compressibility, making it an excellent carrier in solid dispersions and tablets. In contrast, while Florite PS?200 also a carrier which is a petaloid calcium silicate with large macro pores and absorbs up to five times its own weight in liquid, offering deep oil/water uptake along with improved tablet hardness, rapid disintegration, and stabilization of APIs within its pore structure Thus, both carriers support identical suspension volumes, but Neusilin excels at high?capacity adsorption and flow, while Florite enhances compressibility and stabilization. Also, formulation of plain ITZ and nano suspension loaded tablets. The nano suspension has been characterized for particle size measurement. The adsorb suspension powder has been characterized for FTIR (Fourier-transform infrared spectroscopy), DSC (Differential scanning calorimetry), XRPD (X-ray powder diffraction), and polarized light microscopy (PLM). The micronized tablet were evaluated for pre and post compression IPQC test.
MATERIALS AND METHODS
MATERIALS
ITZ was procured from Nifty lab. Kollidon® CL, Soluplus®, and Kollidon® VA 64 was obtained from BASF India Limited, L-HPC was provided by Shin-Etsu Chemical Co., Ltd, India as gift sample. Magnesium stearate was supplied by Merck. Avicel® PH – 102 was gifted by Nitika Pharma. Neusilin UFL 2 and Florite PS -200 was provided by Fuji chemical. Tween 80 was provided by Molychem Pharma.
METHODS
Preparation of Nanosuspension using IKA Homogeniser
A 250 mL suspension was prepared by first forming the aqueous phase using distilled water as the vehicle. To this, a polymer (1–5%), a surfactant (1–5%), and the drug (5–15%) were added in varying concentrations and mixed thoroughly. The mixture was initially homogenized using an overhead stirrer (REME Elektrotechnik LTD) at 1000 RPM for 15 minutes. Subsequently, particle size reduction was carried out using an IKA T18 digital Ultra-Turrax homogenizer at varying speeds of 3000, 5000, 7000, and 10000 RPM for time intervals of 15, 30, and 60 minutes. Results indicated that increasing RPM effectively reduced particle size, whereas extending the homogenization time had minimal additional impact. Using this process, the particle size of itraconazole (ITR) was successfully reduced from 1200 µm to approximately 25 µm. The resulting suspension was then further processed using a high-pressure homogenizer to achieve nanometer-sized particles.
Preparation of Nanosuspension using High Pressure Homogeniser
The micronized suspension obtained from the IKA homogenizer was further processed using a High-Pressure Homogenizer (GEA Niro Soavi Panda Plus) operated at a pressure range of 7000–8000 psi. Samples were collected at specific time intervals (15, 30, and 45 minutes) to monitor particle size reduction. Particle size measurements were performed using a Zeta Sizer (Mastersizer 2000). This process effectively reduced the desired particle size of Itraconazole (ITR). In this study, four suspension formulations (B1–B4) of Itraconazole (ITZ) by using different polymers and surfactant concentrations. Batch B1 contained 5% ITZ with HPMC E5 (1%) and Tween 80 (1%) in distilled water, while batches B2–B4 contained 10% ITZ with polymer (5%) and surfactant (5%) concentrations. B1 and B2 used HPMC E5, B3 used Kollidon VA64, and B4 used Soluplus as polymer (Table 1).
Table 1 Formulation of suspension
|
Formulation of suspension |
|||||
|
% W/W |
|||||
|
Sr.no |
Batch |
Drug (ITZ) |
Polymer |
Surfactant (Tween 80) |
Distilled water |
|
1 |
B1 |
5% |
HPMC E5 (1%) |
1% |
93% |
|
2 |
B2 |
10% |
HPMC E5 (5%) |
5% |
80 % |
|
3 |
B3 |
10% |
Kollidon VA 64 (5%) |
5% |
80 % |
|
4 |
B4 |
10% |
Soluplus (5%) |
5% |
80 % |
Adsorption of Nanosuspension using Carrier
Neusilin UFL2 and Florite PS?200 both effectively used as a carrier to convert liquid suspensions batch (B3/B4) into free-flowing powders. Initially, weighed 10?g of carrier like Neusilin UFL?2 and Florite PS?200 add nano suspension (batch B3 or B4) on it. To this add 35?mL of the suspension on the carrier and sample at 50?°C for 1?hour using hot-air oven (SIENA instrument, Mumbai). Further, repeat same process again twice and dried again at 50?°C for 1?hour. After three cycles, by adsorbing 105?mL per 250?mL batch, but their adsorption strengths and physical characteristic. The final product was 10.5?g of drug-loaded free flowing powder, demonstrating nearly complete adsorption of the nanosuspension.
Table 2 Adsorption of Nanosuspension Using Carrier
|
Suspension Batch |
Adsorption Carrier |
Adsorption Code |
Suspension Volume (mL) |
|
B3 |
Neusilin UFL2 |
A1 |
105 |
|
B3 |
Florite PS200 |
A2 |
105 |
|
B4 |
Neusilin UFL2 |
A3 |
105 |
|
B4 |
Florite PS200 |
A4 |
105 |
Formulation of Immediate Release Tablet
Initially, weigh accurately equivalent 100 mg adsorb powder and add other tablet excipients mixed thoroughly and pass through #40 mesh sieve and to get uniform size. Further, add glidant and lubricant and mix thoroughly. Punch the tablets by using concave punch no 10 mm on tablet compression machine (Cadmac Machinery Co. PVT. LTD) during this the weight and hardness optimised. Kollidon?VA?64 is a dry, direct-compression binder that also enhances solubility and forms instant-release matrices , Soluplus similarly solubilizes poorly water-soluble drugs and stabilizes amorphous dispersions , MCC PH?102 provides excellent flow and compatibility, L?HPC swells rapidly for quick disintegration, Kollidon?CL promotes tablet breakup (disintegration), and magnesium stearate ensures smooth ejection. Together, these excipients create tablets that are robust, easy to manufacture, rapidly disintegrating, and effective at drug release.
Table 3 Formulation of Immediate release tablet
|
|
A1 |
A2 |
A3 |
A4 |
||||
|
Ingredients |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
|
DRUG: Kollidon VA 64/Soluplus |
160 |
160 |
160 |
160 |
160 |
160 |
160 |
160 |
|
MCC PH 102 |
124 |
124 |
124 |
124 |
120 |
120 |
120 |
120 |
|
L –HPC |
35 |
35 |
35 |
35 |
35 |
35 |
35 |
35 |
|
Kollidon – CL |
24.5 |
24.5 |
24.5 |
24.5 |
28 |
28 |
28 |
28 |
|
Magnesium stearate |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
Total |
350 mg |
|||||||
Tablet Evaluation Test
The precompression studies like powder flow properties includes bulk density, tapped density, carr’s index, hausner’s ratio, angle of repose were carried out (DBK instruments, Mumbai). While the post compression study includes drug content, weight variation, disintegration time (DBK instruments, Mumbai), Friability (Roche friabilator, DBK instruments, Mumbai), hardness (ORCHID tester), thickness (vernier calliper), and in-vitro dissolution (USP basket type dissolution test apparatus (LAB INDIA).
RESULT AND DISCUSSION
Particle Size Measurement
The IKA homogenizer dramatically reduced the initial ~1200?µm drug particles to ~23–31?µm by applying intense mechanical shear through a rapidly spinning rotor in a stator at 3000–7000?RPM over 30?minutes. Specifically, increasing the speed from 3000?RPM (B1: 30.69?µm) to 7000?RPM (B3: 23.03?µm) achieved the smallest particles. This outcome aligns with typical high-shear rotor–stator performance, which commonly attains particle sizes in the 2–5?µm range, though reaching ~10?µm is more usual in standard operations. When the 10% drug nanosuspension B3 and B4 were processed using a high-pressure homogenizer at 7000–8000?psi, the particle size decreased significantly over time: it started around 1,300?nm after 15 minutes, halved to roughly 600–676?nm after 30 minutes, and reached a final size of about 565–595?nm at 45 minutes. This clear trend illustrates how high pressure, combined with shear forces and cavitation, drives efficient nanoscale size reduction in suspensions.
Table 4 Particle size measurement
|
IKA homogenizer |
|||||
|
Sr.no |
Sample (250 ml) |
Drug loading |
RPM |
Time interval (min) |
Particle size in (um) |
|
1. |
Plain Drug |
- |
- |
- |
1203.10 um |
|
2. |
B1 |
5 % |
3000 |
30 |
30.69 um |
|
3. |
B2 |
10 % |
5000 |
30 |
29.52 um |
|
4. |
B3 |
10 % |
7000 |
30 |
23.03 um |
|
5. |
B4 |
10 % |
7000 |
30 |
24.73 um |
|
High pressure homogenizer |
|||||
|
Sr.no |
Sample (250 ml) |
Drug loading |
Pressure (psi) |
Time interval (min) |
Particle size in (nm) |
|
1. |
B3 |
10 % |
7000-8000 |
15 min |
1298 nm |
|
2. |
B3 |
10 % |
7000-8000 |
30 min |
676 nm |
|
3. |
B3 |
10 % |
7000-8000 |
45 min |
595 nm |
|
5. |
B4 |
10 % |
7000-8000 |
15 min |
1228 nm |
|
6. |
B4 |
10 % |
7000-8000 |
30 min |
592 nm |
|
7. |
B4 |
10 % |
7000-8000 |
45 min |
565 nm |
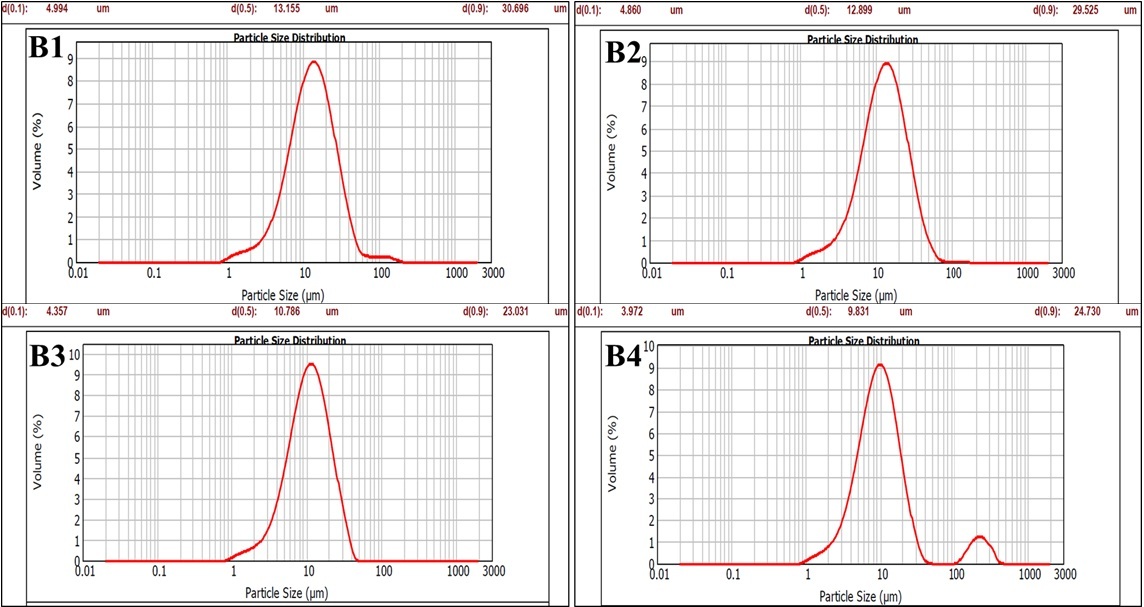
Figure 1 Particle Size Measurement (IKA homogenizer) |
|
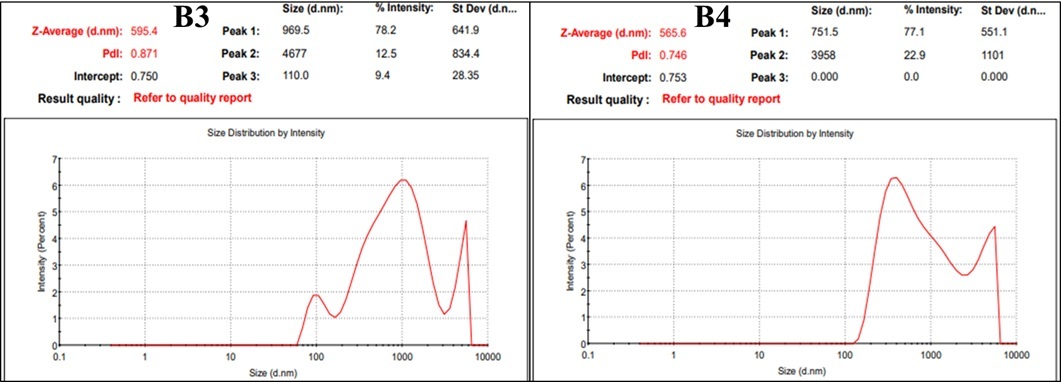
Figure 2 Particle Size Measurement (HPH) |
Drug Content
The four batches (A1–A4) show drug contents of 96.11%, 92.05%, 91.42%, and 94.70%, respectively, averaging approximately 93.07% of the label claim comfortably within the pharmacopeial acceptance criteria of 90–110%.
Polarized Light Microscope (PLM)
Investigation of the samples were done by examination under polarized light microscopy, in order to study the texture of the anisotropic phases. A small quantity of the sample was placed on a clean glass slide. The existence of birefringence was verified by observation under crossed polar employing magnification of 10X. Photomicrographs of these samples were
Itraconazole shows irregular, somewhat coarse particles. Neusilin UFL2 exhibits a highly porous, fine, agglomerated sub?micron structure, consistent with literature describing its large surface area and porosity while Florite PS 200 a porous silicate, displays more uniform, small granules ideal for adsorbing liquids. The A1 and A3 show sizable dense clusters, indicating substantial binding or aggregation likely reflective of high drug: excipient loading or strong agglomeration. Also, A2 appears as a dispersed powder, suggesting better surface coverage by the excipient and more uniform distribution. The A4 shows finely distributed smaller particles, implying superior deagglomeration or wetting.

Figure 3 Polarized Light Microscope (PLM) |
Fourier-transform infrared spectroscopy (ATR-FTIR)
FTIR studies were any interactions between itraconazole (ITZ) and the excipients used in formulations A1 to A4. In formulations A1 and A2 (Kollidon VA64 with Neusilin UFL2 and Florite PS200) showed the same ITZ peaks shows characteristic peaks between 1600–500 cm?¹, particularly in regions around 1500–1000 cm?¹, which correspond to aromatic C=C stretching, C–O stretching, C–N vibrations, kollidon VA64 has its own distinct peaks, especially C=O and C–N vibration, but with slight changes in their position and intensity. This suggests some physical interaction like hydrogen bonding between the drug and the polymer. However, no new peaks appeared, which means there was no chemical reaction or breakdown of the drug. While formulations A3 and A4 (using Soluplus and carrier) also show broadening and slight shifting of ITZ peaks between 1500–500 cm?¹. The peaks of ITZ were still present, but with a bit more shift and broadening, especially compared to Kollidon. This describes stronger interaction between ITZ and Soluplus, but again, the drug remained chemically stable. Therefore, it confirms that ITZ is compatible with all the excipients used. Only physical interactions occurred, which can help improve solubility and stability of the drug, without changing its structure.
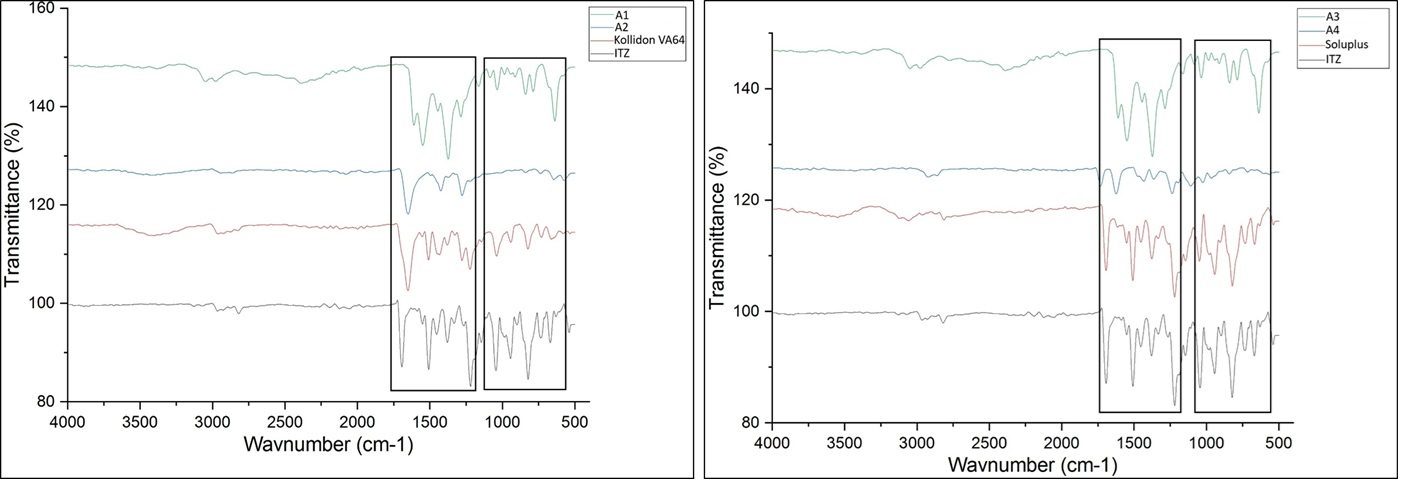
Figure 4 Fourier-Transform Infrared Spectroscopy (ATR-FTIR)
Differential Scanning Calorimetry (DSC)
DSC studies were performed to analyse the thermal behavior and physical state of itraconazole (ITZ) and the polymers used—Kollidon VA64 and Soluplus. The thermogram of pure Itraconazole showed a sharp endothermic peak at around 165°C, corresponding to its melting point. This sharp peak indicates the crystalline nature of pure ITZ. For Kollidon VA64, multiple broad thermal events were observed in the range of 100°C to 300°C, but no sharp melting peak was seen. This is expected, as Kollidon VA64 is an amorphous polymer, and its thermogram mainly reflects glass transition and decomposition events, rather than a defined melting point. In the case of Soluplus, the thermogram also showed broad endothermic transitions between 130°C and 220°C, again indicating an amorphous nature with no distinct melting peak. The broad transitions suggest a glass transition temperature (Tg) and some polymer softening or thermal degradation. The presence of the sharp melting peak of ITZ in its pure form and the absence of such a peak in the polymers confirms that ITZ is crystalline, while both Kollidon VA64 and Soluplus are amorphous.
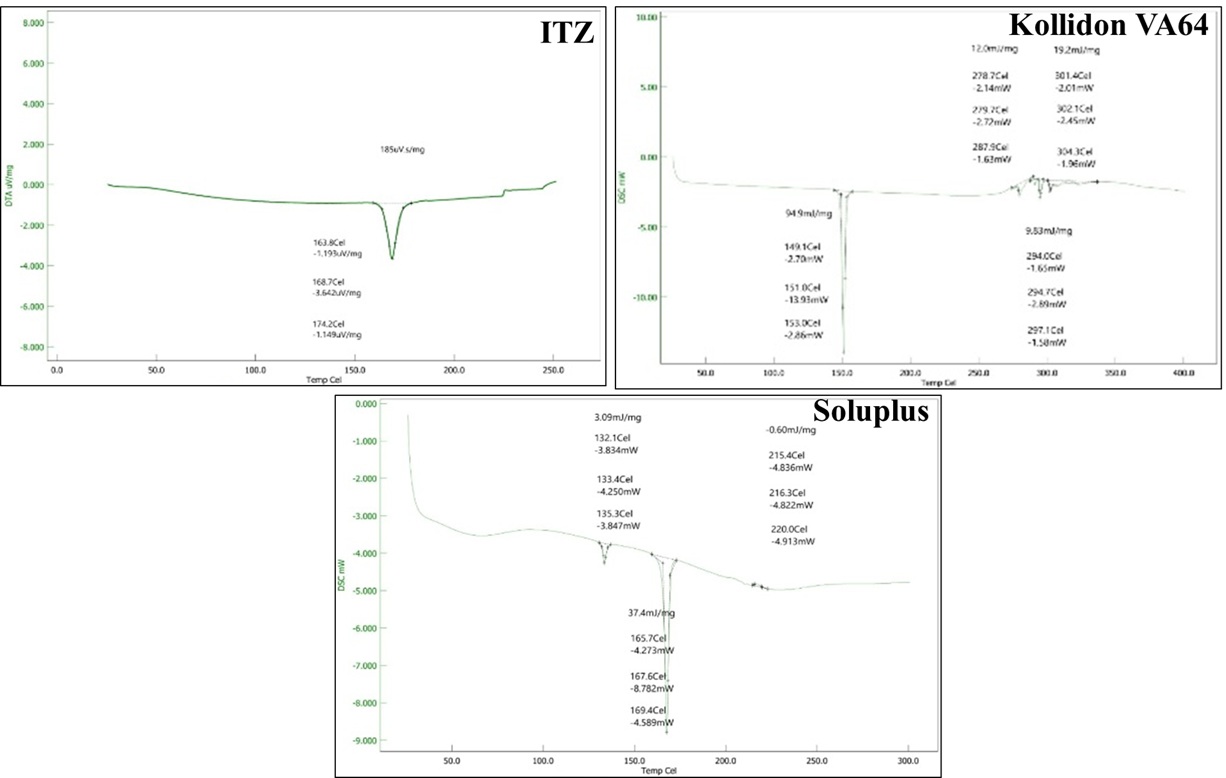
Figure 5 Differential Scanning Calorimetry (DSC) of ITZ, Kollidon VA64 and Soluplus
DSC thermograms were recorded for formulations A1 to A4 to investigate the thermal behavior and physical state of itraconazole (ITZ) in the prepared systems. In the pure drug, a sharp endothermic peak was previously observed around 185°C, which corresponds to the melting point of crystalline itraconazole. In formulation A1, a broad thermal event was observed, and the intensity of the ITZ melting peak around 185°C was significantly reduced and shifted, indicating a possible reduction in crystallinity. The appearance of new or modified thermal transitions between 140°C to 170°C suggests molecular dispersion of ITZ in the excipients, likely due to interaction with Kollidon VA64 and Neusilin UFL2. While A2, containing Florite PS200 and Kollidon VA64, also showed a broad and less intense endothermic region between 160–180°C, with additional minor transitions at lower temperatures. This suggests that ITZ is partially converted into an amorphous state or entrapped in the polymer matrix, which disrupts its crystalline structure. Similarly, A3 showed endothermic peaks between 150–185°C, but the peaks were less sharp and more diffused compared to pure ITZ. This again confirms a reduction in drug crystallinity, likely due to interaction with Soluplus and Neusilin UFL2, which may help in enhancing solubility and stability. In A4, the DSC curve showed a broad thermal transition and absence of a sharp melting point peak of ITZ. Peaks in the range of 155–168°C were observed but were less intense. This suggests that the drug is mostly in an amorphous or molecularly dispersed form, likely stabilized by Soluplus and Florite PS200.
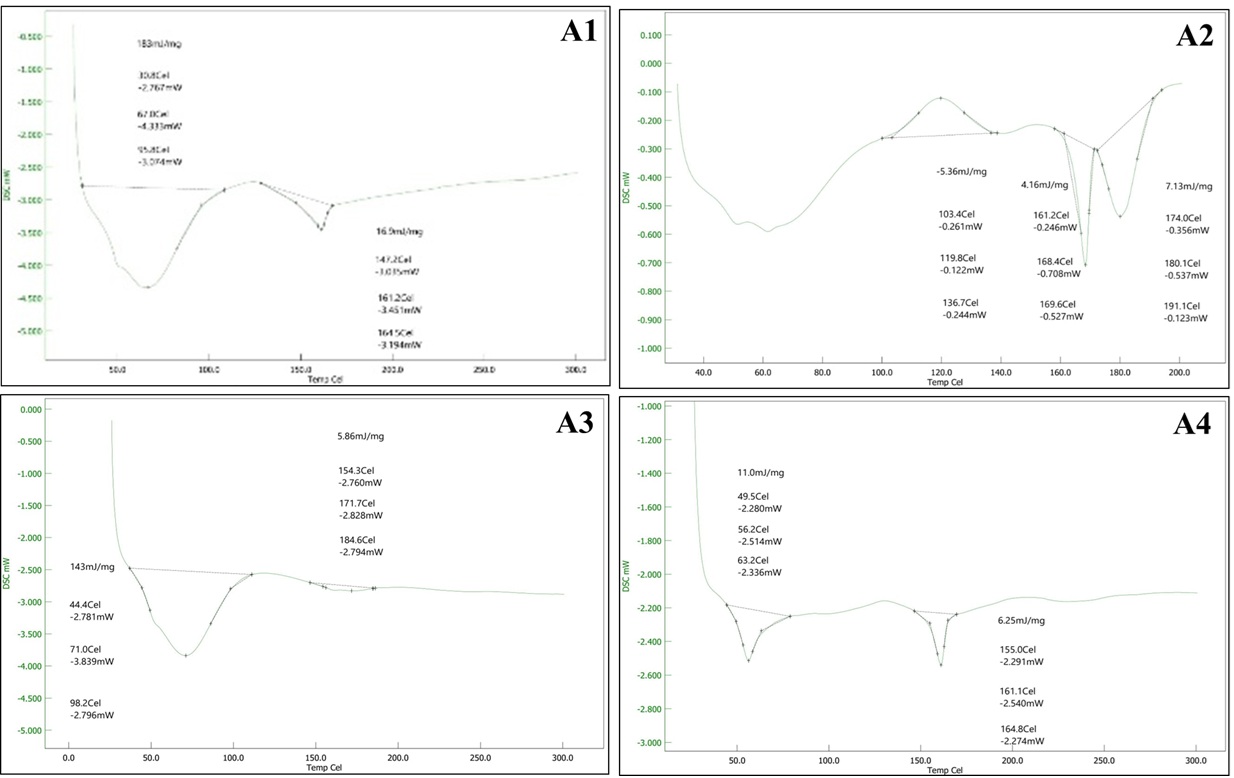
Figure 6 Differential Scanning Calorimetry (DSC) of A1, A2, A3 and A4
X-ray powder diffraction (PXRD)
X-ray diffraction (XRD) was used to evaluate the crystalline or amorphous nature of itraconazole and the polymers used in formulation. The XRD pattern of pure ITZ shows multiple sharp, intense peaks, especially in the range of 10° to 30° (2θ). These sharp peaks are characteristic of a highly crystalline structure, confirming that the drug exists in a well-ordered crystalline form in its pure state. In contrast, kollidon VA64 displays a broad hump or halo without any sharp diffraction peaks. This indicates it is completely amorphous, lacking long-range molecular order. Similarly, the soluplus also exhibits a broad diffuse pattern, confirming its amorphous nature.
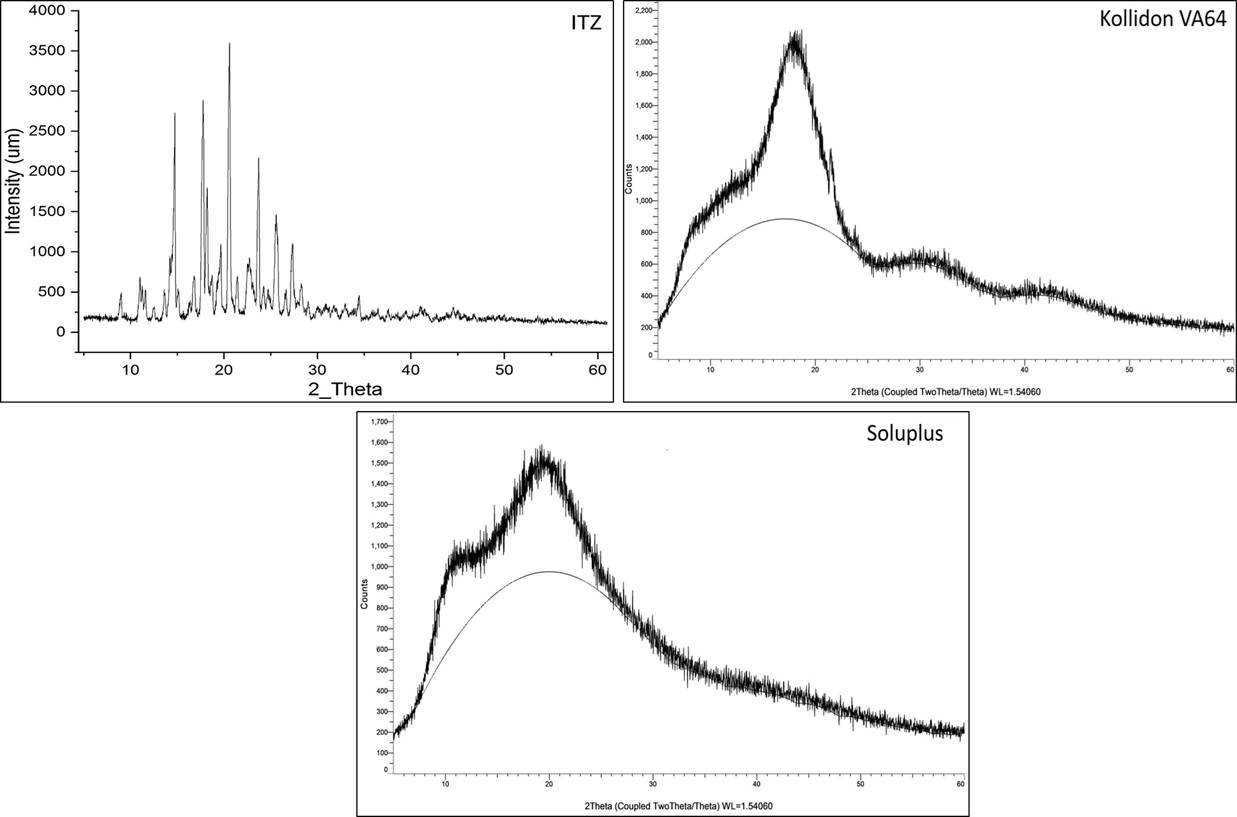
Figure 7 X-ray Powder Diffraction (PXRD)
X-ray diffraction (XRD) was carried out to evaluate the physical state of itraconazole (ITZ) in formulations A1 to A4 and to determine whether it remained crystalline or was converted to an amorphous form upon formulation. In A1 and A3, the XRD patterns show multiple sharp and intense peaks, especially in the 10°–30° (2θ) range, which are similar to those observed in the pure crystalline form of ITZ. This indicates that ITZ remains partially crystalline in these formulations, even after being adsorbed onto Neusilin UFL2 and combined with either Kollidon VA64 (A1) or Soluplus (A3). However, compared to the pure drug, the intensity of the peaks is reduced, suggesting some reduction in crystallinity and possible partial amorphization. In contrast, A2 (with Florite PS200 and Kollidon VA64) shows a broad halo pattern with no sharp peaks, which is a clear indication of amorphous conversion of itraconazole. The absence of distinct crystalline peaks suggests that ITZ has been well incorporated into the polymer matrix in an amorphous or molecularly dispersed state, which is favorable for improving solubility and dissolution. Similarly, A4 (with Florite PS200 and Soluplus) shows a pattern with significantly reduced peak intensity, and the crystalline peaks are almost absent. This again suggests that most of the crystalline ITZ has been transformed into an amorphous form, likely due to the combined effect of Soluplus and the porous carrier Florite PS200. The XRD results demonstrate that formulations A2 and A4 have successfully converted ITZ into an amorphous state, while A1 and A3 still retain some crystalline content. Amorphous conversion is important as it can lead to enhanced solubility and faster dissolution, which is beneficial for improving the oral bioavailability of poorly water-soluble drugs like itraconazole.
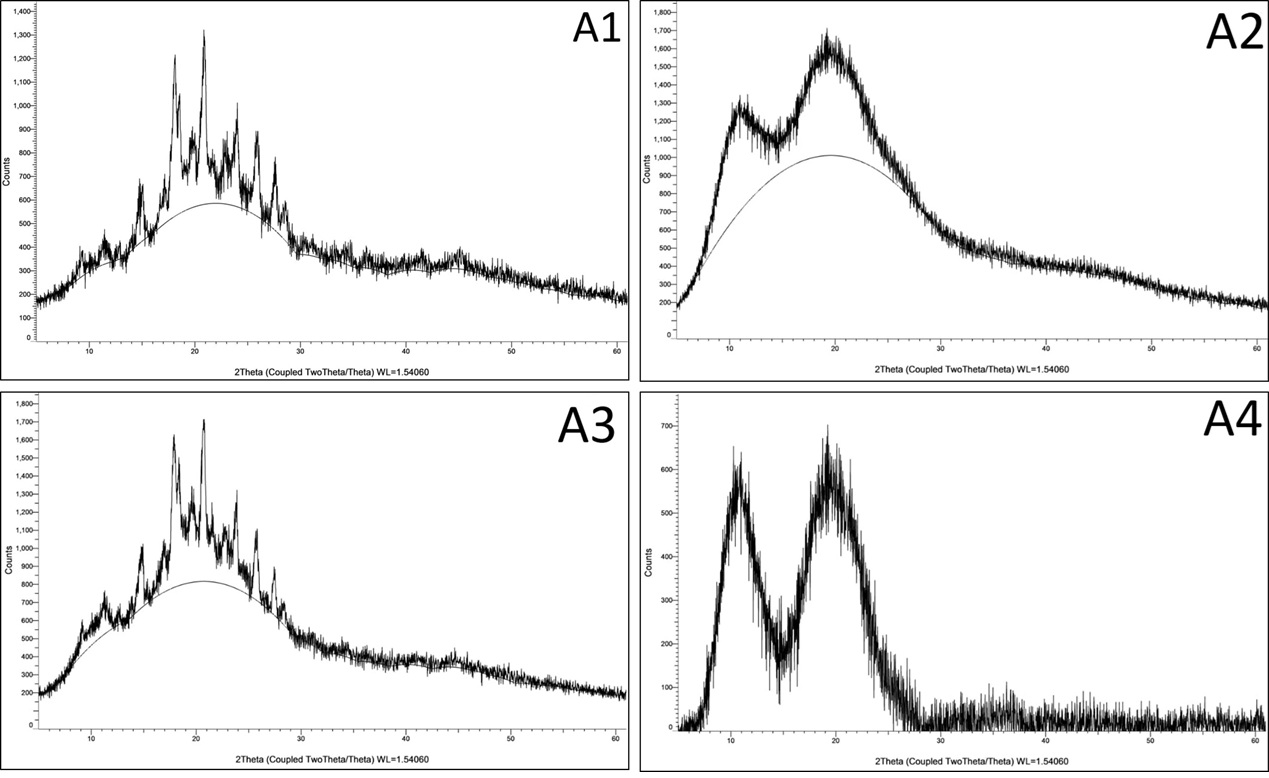
Figure 8 X-ray powder diffraction (PXRD)
Tablet Evaluation Test
Precompression Evaluation
The precompression evaluation of all tablet formulation excipient shown excellent flow properties as compared to the pure drug the compressibility index shown the good flow properties. The pure drug exhibits very poor flow properties low bulk density (0.238?g/mL), high Carr’s index (48.7%), high Hausner’s ratio (1.96), and an unmeasurable angle of repose—indicating extreme cohesion and no free-flowing behaviour. In contrast, all formulated blends (F1–F8) markedly improved flow: bulk and tapped densities increased (0.32–0.50?g/mL and 0.38–0.59?g/mL respectively), yielding Carr’s indices mostly between ~10–15% and Hausner’s ratios of ~1.11–1.18, which fall into “excellent” to “good” flow categories. Their angles of repose (12–22°) further confirm excellent to fair flow ability. These results align well with standard classification thresholds Carr’s index <10% and Hausner’s ratio <1.11 indicate excellent flow, 11–15%/1.12–1.18 indicate good flow, and 16–20%/1.19–1.25 denote fair flow demonstrating that formulation successfully transforms a cohesive API into free?flowing blends suitable for efficient tableting or encapsulation
Post Compression Evaluation
Disintegration Time
The disintegration time of the all the tablet formulation (F1 to F8) was found to be in the limit. The immediate release tablet official disintegration time 30 min and all the tablet formulation show the disintegration time below 30 min hence this test is pass on the basis of lowest disintegration time of formulation and drug content The drug assay results for the optimized formulation (F1 to F8) ranged from 99.21% to 105% range.
Friability
The friability of the tablet formulation (F1 to F8) was found to be in range of (0.55 - 0.74 %) and the official limit of friability is less than 1 % then test was pass.
Weight Variation
The weights of 20 individual tablets formulation (F1 to F8) average around 345–350?mg with a standard deviation of ±5?mg, showing a variation of about ±5% relative to the mean. According to USP weight uniformity guidelines, tablets weighing more than 250?mg may vary by up to ±5% (i.e., no more than 5% deviation from the average) to comply Since all individual tablet weights fall within approximately ±5% of the mean and the standard deviation remains moderate, the batch clearly meets pharmacopeial criteria for weight variation, indicating consistent dosing potential and reliable manufacturing control all tablet formulation was pass the weight variation test of tablet.
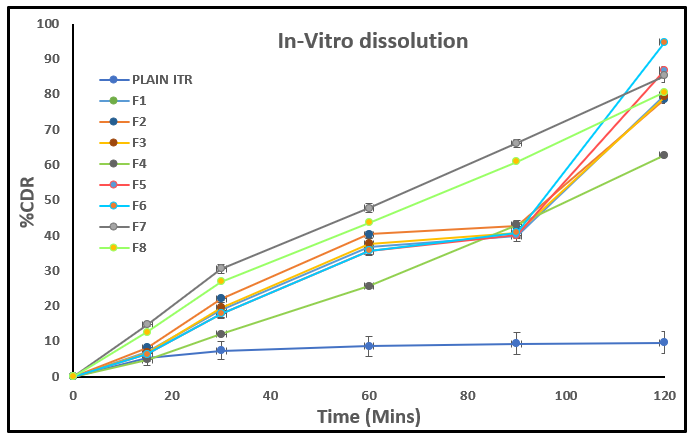
In-vitro Dissolution
The in vitro drug release study at 120 minutes clearly shows that the plain Itraconazole (ITZ) exhibited, with only 9.64% ± 3.17 drug release, indicating its poor aqueous solubility. In contrast, all the developed formulations (F1 to F8) demonstrated a significant improvement in drug release, confirming successful enhancement of solubility and dissolution rate. Among these, F6 showed the highest release (94.7% ± 0.69), followed by F5 (86.74% ± 2.43) and F7 (85.19% ± 1.82). Other formulations like F1, F2, F3, and F8 also showed good release profiles (around 78–80%), while F4 had a relatively lower release (62.77% ± 0.67). These results suggest that the developed formulations, especially F6, significantly improved the release of itraconazole, likely due to the conversion into an amorphous state, use of solubilizing polymers, and optimized carriers.
CONCLUSION
In summary, this study demonstrated the successful formulation of an itraconazole nanosuspension using high-pressure homogenization, with Tween 80 as a stabilizer and polymers such as HPMC E5, Kollidon VA64, and Soluplus as solubility enhancement polymer. The nanosuspension was adsorbed onto Neusilin UFL2 and Florite PS200 to produce a free-flowing powder suitable for tablet formulation. The optimized system exhibited a mean particle size of approximately 500 nm, for achieving this particle size some process parameter was affected like drug loading, RPM and Pressure were investigated and optimized to produce the smallest drug nanoparticles. High drug content, and good stability, while maintaining the crystalline nature of the drug. Tablet formulations passed all standard quality control evaluations. Notably, the in vitro dissolution profile in 0.1N HCl showed a significant enhancement in drug release compared to pure itraconazole, confirming that the nanosuspension approach is a promising strategy for improving the solubility and potential oral bioavailability of poorly water-soluble drugs.
REFERENCES



Sukesh Padamwar*, Ritik Ramdhani, Dr. Arun Mahale*, Micronization-Assisted Solubility Enhancement and Formulation of Immediate Release Tablets for A Poorly Soluble Drug Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 587-603. https://doi.org/10.5281/zenodo.16751633
 10.5281/zenodo.16751633
10.5281/zenodo.16751633
