
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Bhagwan Mahavir College Of Pharmacy, BMEF Campus, Bharthana, Vesu, Surat 395017
Biologics have transformed the treatment of chronic and life-threatening diseases, but their high cost remains a barrier to equitable access worldwide. Biosimilars—highly similar versions of approved biologics—offer a pathway to cost reduction and improved healthcare access. Over the last decade, and particularly in 2024–2025, biosimilars have experienced rapid regulatory, clinical, and market expansion. This review critically examines the scientific foundations of biosimilarity, regulatory advances, clinical evidence of safety and efficacy, and market adoption trends, with a special focus on India. Analytical comparability, pharmacokinetics, immunogenicity, and switching studies consistently demonstrate no clinically meaningful differences between biosimilars and their reference biologics. The European Medicines Agency (EMA) and U.S. FDA have advanced regulatory science by recognizing interchangeability and refining requirements, while India has emerged as a leader among developing nations, approving more than 120 biosimilars. Market data highlight substantial price erosion—ranging from 20% to 70%—across therapeutic areas, improving patient access to oncology, autoimmune, and diabetes treatments. Pharmacovigilance systems, including India’s PvPI, ensure long-term safety monitoring. Despite barriers such as prescriber hesitation and limited awareness, biosimilars are positioned to reshape global healthcare systems by 2030. This article synthesizes data, real-world evidence, and regulatory milestones to provide a comprehensive perspective on the evolving landscape of biosimilars.
Biological medicines, or biologics, are therapeutic products derived from living systems such as mammalian cells, yeast, or bacteria. They include monoclonal antibodies, recombinant hormones, vaccines, and therapeutic proteins, all of which have transformed modern medicine [1]. Biologics are now the backbone of therapy for cancer, autoimmune diseases, diabetes, and rare genetic disorders. However, the high cost of biologics limits their accessibility. In high-income countries, annual treatment costs for monoclonal antibodies such as trastuzumab or adalimumab range from USD 20,000 to 70,000 per patient [2]. In India, although prices are lower, they still remain unaffordable for many patients, where out-of-pocket spending accounts for nearly 60% of healthcare expenditure [3].
To address these challenges, biosimilars—biological medicines that are highly similar to approved reference products—have been developed. Unlike small-molecule generics, biosimilars cannot be exact copies due to the complex structures and manufacturing processes involved. Instead, they undergo a rigorous comparability exercise, involving detailed analytical characterization, pharmacokinetics (PK), pharmacodynamics (PD), immunogenicity, and confirmatory clinical studies [4].
Globally, the biosimilar market has grown exponentially since the first approval in Europe in 2006. As of 2025, the EMA has approved more than 90 biosimilars, the U.S. FDA has approved over 45, and India has licensed more than 120, making it a frontrunner in the developing world [5,6]. This expansion is expected to propel the global biosimilar market beyond USD 100 billion by 2030 [7].
Figure 1.1: Global biosimilar approvals by EMA, FDA, and India (2006–2025).
India has played a crucial role in biosimilar development. The Central Drugs Standard Control Organization (CDSCO) and Department of Biotechnology (DBT) issued the first biosimilar guidelines in 2012, updated in 2016, with a revised draft released in 2025 [8]. These guidelines ensure rigorous evaluation while balancing patient access. Indian companies such as Biocon, Intas, Dr. Reddy’s Laboratories, and Zydus Cadila have emerged as global players, exporting biosimilars to over 70 countries [9].
The objective of this review is to provide a comprehensive understanding of the safety, efficacy, and regulatory landscape of biosimilars in 2024–2025. The article will cover:
By integrating data, facts, figures, and real-world examples, this article seeks to highlight the transformative role biosimilars can play in addressing healthcare affordability while maintaining patient safety and clinical effectiveness.
2. Scientific Basis of Biosimilarity
The foundation of biosimilar development lies in establishing high similarity to the reference biologic without clinically meaningful differences in quality, safety, or efficacy. This principle, known as the totality-of-evidence approach, requires integration of analytical, preclinical, pharmacokinetic/pharmacodynamic (PK/PD), and clinical data [10]. The following subsections outline the scientific processes involved.
Analytical characterization is the most critical step in biosimilar development. Modern technologies allow detailed assessment of structural and functional attributes.
Table 2.1: Analytical comparability methods in biosimilar evaluation.
|
Category |
Techniques |
Purpose |
|
Primary Structure |
Peptide mapping (LC-MS/MS), Edman sequencing |
Confirms amino acid sequence identity |
|
Higher Order Structure |
Circular dichroism (CD), FTIR, NMR, X-ray crystallography |
Verifies secondary/ tertiary structure equivalence |
|
Post-Translational Modifications (PTMs) |
Glycan profiling (HPLC, LC-MS), CE-SDS, isoelectric focusing (IEF) |
Detects glycosylation, charge heterogeneity, phosphorylation, oxidation |
|
Purity & Impurities |
SEC-HPLC, CE-SDS, RP-HPLC, AUC |
Quantifies aggregates, fragments, degradation products |
|
Biological Activity |
Cell-based assays, binding affinity assays (SPR, ELISA, BLI) |
Confirms mechanism of action (MoA) and receptor binding |
|
Stability |
Stress testing (thermal, oxidative, pH, light), accelerated degradation studies |
Compares stability profile under forced conditions |
Biosimilars must demonstrate PK/PD equivalence to the reference product. Typically, healthy volunteer studies are performed to compare:
Cmax (peak concentration) and AUC (area under the curve)
Equivalence is achieved if the 90% confidence interval of the ratio of biosimilar to reference falls within 80–125% [16]. For biologics with complex dynamics, validated PD markers such as neutrophil counts (for G-CSF products) or blood glucose levels (for insulin analogues) are critical [17].
Unlike generics, biosimilars require at least one confirmatory clinical trial to assess efficacy and safety in a sensitive indication. These trials focus primarily on immunogenicity, i.e., the formation of anti-drug antibodies (ADA) and neutralizing antibodies (NAb) [18].
For example, in the PLANETRA trial (infliximab biosimilar), the incidence of ADA was comparable between the biosimilar and reference product [19].
Immunogenicity is influenced by manufacturing processes, glycosylation patterns, and even container closure systems [20].
If biosimilarity is demonstrated in one clinical indication, regulators may permit extrapolation to all approved indications of the reference biologic. This is based on scientific justification of mechanism of action, receptor binding, and pharmacology [21].
For instance, adalimumab biosimilars were initially studied in rheumatoid arthritis but subsequently approved for psoriasis, Crohn’s disease, and ulcerative colitis [22].
In India, extrapolation has been accepted for several monoclonal antibodies, provided analytical and PK/PD data support similarity [23].
Trastuzumab (Herceptin® biosimilars): Extensive analytical comparability and clinical trials in HER2-positive breast cancer confirmed biosimilarity, leading to approvals across multiple countries [24].
Insulin glargine (Semglee®): Demonstrated PK/PD equivalence and clinical efficacy, and became the first interchangeable biosimilar insulin approved by the U.S. FDA [25].
Etanercept (Erelzi® biosimilars): Confirmatory clinical trials in psoriasis supported extrapolation to multiple autoimmune conditions [26].
2.6 Significance of Scientific Evaluation
The scientific rigor behind biosimilarity ensures that patients and prescribers can trust these products to deliver the same clinical benefit as reference biologics. Importantly, over 15 years of global experience has yielded no unexpected safety concerns, reinforcing the validity of this stepwise approach [27].
3. Global Regulatory Frameworks
Biosimilars represent a regulatory challenge because, unlike generics, they are similar but not identical to their reference biologics. Regulatory authorities worldwide have therefore established science-based pathways to ensure that biosimilars meet rigorous standards of quality, safety, and efficacy. This section highlights the frameworks of the European Union (EU), the United States (US), the World Health Organization (WHO), and India, with a comparative analysis of their approaches.
The European Union (EU) was the first jurisdiction to introduce a legal framework for biosimilars in 2004, with the first approval granted in 2006 for Omnitrope® (somatropin) [28].
Guidelines: EMA established class-specific guidelines (e.g., for monoclonal antibodies, insulins, erythropoietins).
Requirements: A comprehensive comparability exercise is required, starting with analytical similarity and extending to PK/PD and confirmatory clinical trials.
Extrapolation: EMA allows extrapolation of indications if scientifically justified.
Interchangeability: In 2022, the EMA and HMA (Heads of Medicines Agencies) confirmed that all biosimilars approved in the EU are considered interchangeable with their reference product [29].
Market penetration: The EU leads globally, with biosimilars accounting for >70% of market share in epoetins and G-CSF products [30].
The US established its pathway for biosimilars under the Biologics Price Competition and Innovation Act (BPCIA) of 2010, creating the 351(k) pathway [31].
First approval: Zarxio® (filgrastim-sndz) in 2015.
Requirements: Stepwise approach, with strong emphasis on analytical similarity, animal studies (where necessary), and at least one clinical study in a sensitive indication.
Purple Book: FDA maintains a database listing all approved biologics and biosimilars, along with interchangeability status [32].
Interchangeability: Biosimilars may receive an interchangeability designation, permitting automatic substitution at the pharmacy level. Examples include Semglee® (insulin glargine), Cyltezo® (adalimumab-adbm), and Cimerli® (ranibizumab) [33].
Recent advances: In 2024, FDA proposed removing mandatory switching studies for interchangeability, signaling regulatory convergence with EMA [34].
The WHO plays a central role in shaping biosimilar regulations for low- and middle-income countries (LMICs).
First guideline: Published in 2009, introducing the concept of Similar Biotherapeutic Products (SBPs) [35].
Revision: Updated in 2022, emphasizing a risk-based, flexible approach, reducing unnecessary clinical studies when strong analytical data exist [36].
Reliance model: WHO encourages regulators in LMICs to rely on EMA/FDA-approved biosimilars to accelerate access.
Impact: Many countries in Asia, Africa, and Latin America have adopted WHO principles in their national guidelines [37].
India has emerged as a global leader in biosimilars, with over 120 approvals by 2025.
First approval: Recombinant human insulin in 2000 [38].
Guidelines:
Market: Indian companies dominate the domestic market and export biosimilars to over 70 countries. Key players include Biocon Biologics, Intas Pharmaceuticals, Dr. Reddy’s, Zydus Cadila, and Lupin [40].
Challenges: Lack of explicit interchangeability designation and limited pharmacovigilance infrastructure remain hurdles [41].
Global Regulatory Frameworks for Biosimilars
Table 3.1: Global regulatory frameworks for biosimilars.
|
Region / Regulator |
Year of First Guideline / Approval |
Key Features |
Recent Updates |
|
European Union (EMA) |
2006 (somatropin, Omnitrope®) |
First biosimilar guideline globally; stepwise comparability (quality, non-clinical, clinical) |
2022: EMA/HMA interchangeability statement |
|
United States (FDA) |
2010 (BPCIA 351(k) pathway) |
Requires analytical + PK/PD comparability; extrapolation allowed; interchangeability designation |
2024 draft: Reduces reliance on switching trials |
|
World Health Organization (WHO) |
2009 (SBP guideline) |
Provides global reference framework for member states; encourages reliance and risk-based evaluation |
2022 revision: streamlined requirements |
|
India (CDSCO/DBT) |
2012 (first guideline), 2016 (revision) |
Requires confirmatory PK/PD & clinical trials; extrapolation permitted; local bridging |
2025 draft: Accepts global reference products; aligns with WHO |
|
Japan (PMDA) |
2009 |
Similar to EMA; strong analytical focus; case-by-case clinical trial requirements |
Ongoing harmonization with ICH |
|
Canada (Health Canada) |
2010 |
Abbreviated pathway based on EMA; extrapolation permitted |
Updated guidance 2019 |
|
China (NMPA) |
2015 |
Initially restrictive, now moving toward EMA-alignment |
Expanded mAb approvals post-2020 |
|
Brazil (ANVISA) |
2010 |
Allows both standalone and comparability-based development |
Updated 2015: risk-based approach |
Key differences include:
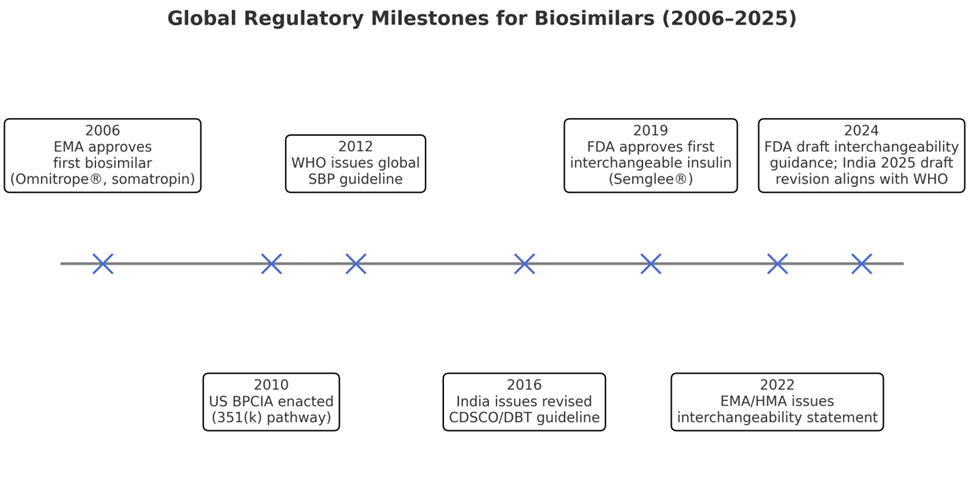
Figure 3.1: Timeline of biosimilar regulatory milestones.
This figure illustrates how global regulatory science has evolved from the EU’s pioneering approvals in 2006 to India’s draft reforms in 2025, highlighting the trend toward harmonization.
4. Clinical Evidence on Safety & Efficacy
The acceptance of biosimilars in clinical practice depends largely on robust evidence demonstrating comparable safety, efficacy, and immunogenicity with their reference biologics. Regulators and clinicians require data from switching trials, meta-analyses, and real-world studies to establish confidence in biosimilar use. Over the last decade, and particularly in the years 2024–2025, this evidence base has expanded substantially.
Switching trials evaluate the outcomes when patients are transitioned from a reference biologic to its biosimilar. These studies focus on efficacy, safety, and immunogenicity after single or multiple switches.
NOR-SWITCH Trial (2017): A randomized trial in Norway involving 482 patients across six indications (rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and chronic plaque psoriasis). The study showed no significant difference in disease worsening between infliximab biosimilar and the originator [42].
EGALITY Trial (2017): A study of 531 psoriasis patients switching multiple times between reference etanercept and its biosimilar. Results confirmed equivalent efficacy, safety, and immunogenicity [43].
VOLTAIRE-X Trial (2021): A phase III study of 238 patients with plaque psoriasis demonstrated that switching between Humira® (adalimumab) and Cyltezo® biosimilar had no impact on efficacy or safety. This trial supported the FDA’s interchangeability designation for Cyltezo® [44].
PLANETRA and PLANETAS Trials: Evaluated infliximab biosimilars in rheumatoid arthritis and ankylosing spondylitis, showing comparable outcomes with reference products [45].
Figure 4.1: Summary of switching trial outcomes.
|
Trial |
Molecule |
Indication(s) |
Patients (n) |
Design |
Outcome |
|
NOR-SWITCH |
Infliximab (CT-P13) |
RA, AS, IBD, PsA, UC, CD |
482 |
Randomized, double-blind switch |
No difference in disease worsening |
|
EGALITY |
Etanercept (SB4) |
Psoriasis |
531 |
Multiple switches |
PASI75, ADA rates equivalent |
|
VOLTAIRE-X |
Adalimumab (BI 695501) |
Psoriasis |
238 |
Multiple switches |
Equivalent efficacy, immunogenicity |
|
PLANETRA/ PLANETAS |
Infliximab (CT-P13) |
RA, AS |
1200+ |
Double-blind switch trials |
Confirmed equivalence |
4.2 Meta-Analyses
Systematic reviews and meta-analyses consolidate data across multiple studies, strengthening the evidence base for biosimilars.
A 2020 meta-analysis covering 90 clinical trials and 14,000 patients concluded that biosimilars had no increased risk of immunogenicity or adverse events compared to reference biologics [46].
Another pooled analysis of switching studies showed no difference in efficacy endpoints (ACR20, PASI75, DAS28 remission rates) after switching [47].
A Cochrane systematic review in 2023 reiterated the clinical equivalence of biosimilars in rheumatology and oncology, supporting broader adoption [48].
Beyond clinical trials, real-world data (RWD) from registries and post-marketing surveillance provide critical insights into long-term safety and effectiveness.
DANBIO Registry (Denmark): Nationwide switch of patients from innovator infliximab to biosimilar infliximab showed stable remission rates and no safety concerns [49].
Swedish Rheumatology Quality Register: Demonstrated comparable persistence rates between etanercept biosimilars and reference products [50].
Indian Oncology Centers: Real-world studies on trastuzumab biosimilars in HER2-positive breast cancer revealed equivalent progression-free and overall survival rates [51].
Pharmacovigilance reports: The European Medicines Agency’s EudraVigilance database shows no new safety signals across 90+ approved biosimilars [52].
Immunogenicity is a major clinical consideration for biosimilars. Anti-drug antibodies (ADA) can reduce efficacy or cause hypersensitivity. However, accumulated evidence indicates comparable immunogenicity rates between biosimilars and reference biologics.
The NOR-SWITCH and EGALITY trials reported no meaningful increase in ADA incidence after switching [53].
Indian clinical studies of rituximab and trastuzumab biosimilars confirmed low and clinically irrelevant ADA rates [54].
4.5 Confidence Building in Clinical Practice
The consistent findings from randomized trials, meta-analyses, and real-world studies have reassured clinicians and regulators that biosimilars can be safely and effectively substituted for reference biologics. However, physician education and patient communication remain essential to increase trust and uptake [55].
5. Market Dynamics & Adoption Trends
The market adoption of biosimilars has been one of the most transformative developments in global healthcare economics. With patent expirations of blockbuster biologics such as adalimumab, trastuzumab, and etanercept, biosimilars have entered the market with the potential to reduce costs, increase patient access, and stimulate competition. This section examines global trends, India-specific developments, price erosion, and barriers to adoption.
Since the first biosimilar approval in 2006, the global market has grown at an annual rate exceeding 25%. According to IQVIA reports, biosimilars generated savings of over €10 billion annually in the European Union alone by 2023 [56].
Europe (EU): Europe remains the largest biosimilar market, with more than 90 approvals by EMA. Market share for certain classes is impressive: epoetins (80%), G-CSF (85%), and infliximab (70%) [57].
United States (US): The U.S. market was initially slow due to regulatory complexity and patent litigation. However, since 2019, biosimilars have gained momentum. By 2025, over 45 products are approved, and adalimumab biosimilars alone captured ~30% of U.S. market share within two years of launch [58].
Asia-Pacific: Countries like South Korea and China have rapidly advanced biosimilar production, focusing on domestic affordability and global exports [59].
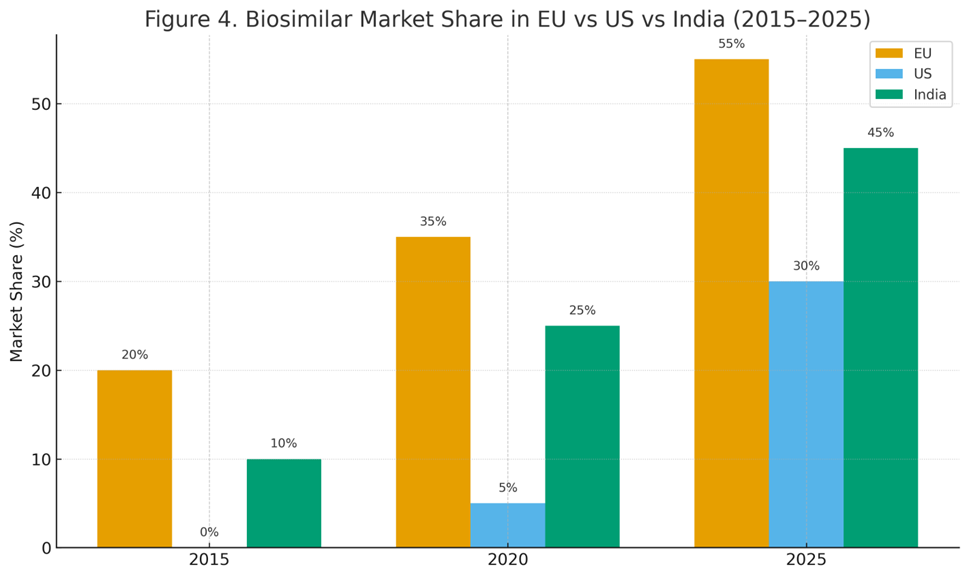
Figure 5.1: Biosimilar market share in EU vs US vs India (2015–2025).
5.2 India-Specific Market
India has become a pioneer in biosimilar development among low- and middle-income countries.
Approvals: By 2025, India has approved 120+ biosimilars, the largest number outside the EU [60].
Market size: Valued at approximately INR 4200 crores (USD 5 billion) in 2025, with a CAGR of 15–18% [61].
Export potential: Indian biosimilars are exported to over 70 countries, including regulated markets such as the EU and the U.S. (through partnerships and co-developments) [62].
Leading companies: Biocon Biologics, Intas Pharmaceuticals, Dr. Reddy’s Laboratories, Zydus Cadila, and Lupin are among the top developers.
Adoption drivers: India’s cost-sensitive market encourages rapid biosimilar uptake, especially in oncology and endocrinology.
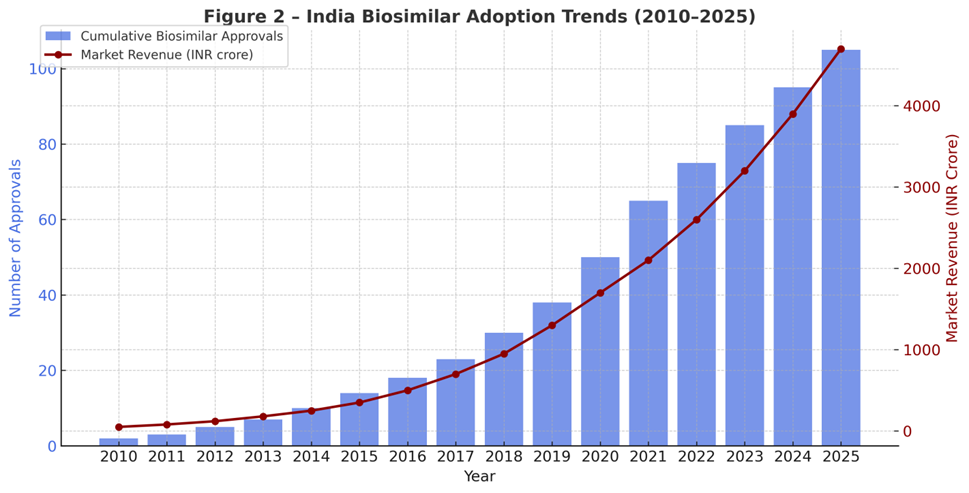
Figure 5.2: India-specific biosimilar adoption trends (approvals and sales revenue, 2010–2025).
5.3 Price Erosion
Biosimilars drive significant price erosion, improving affordability.
In the EU, tender-based procurement has resulted in 50–70% price reductions for epoetins, G-CSF, and monoclonal antibodies [63].
In the US, price erosion averages 20–35%, though competition for adalimumab biosimilars is pushing discounts closer to 50% [64].
In India, trastuzumab and insulin glargine biosimilars are priced 30–40% lower than reference products, expanding access for middle-class and rural populations [65].
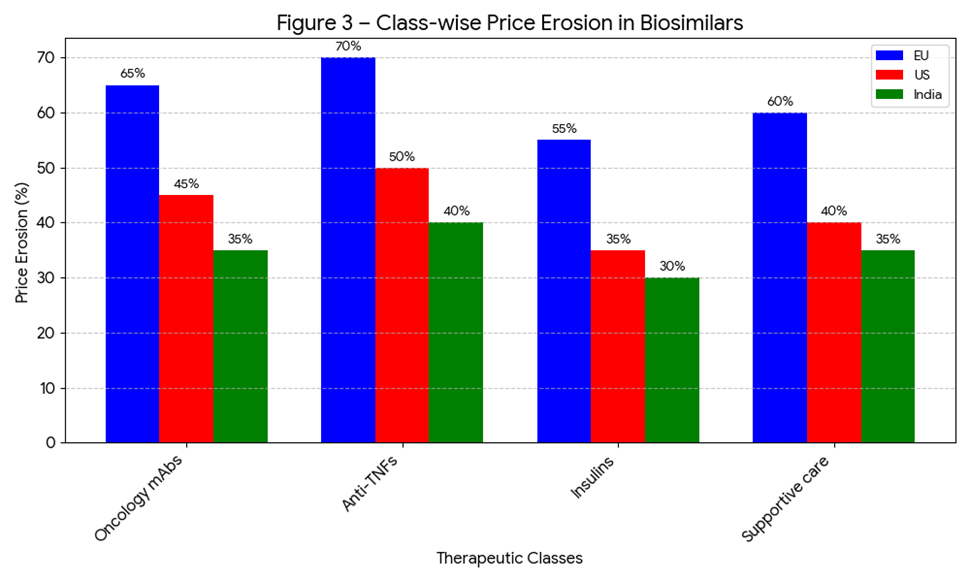
Figure 5.3: Average price erosion by therapeutic class (oncology, diabetes, autoimmune, supportive care).
5.4 Barriers to Adoption
Despite proven equivalence, biosimilars face adoption challenges:
5.5 Case Examples
Adalimumab (Humira®): In the EU, biosimilar entry reduced prices by nearly 70%. In the U.S., within two years of biosimilar entry (2023–2025), originator sales dropped sharply as multiple biosimilars entered the market [70].
Trastuzumab (Herceptin®): In India, the introduction of trastuzumab biosimilars reduced treatment costs for HER2+ breast cancer patients by 30–35%, increasing accessibility in public hospitals [71].
Insulin Glargine: Semglee® became the first FDA-approved interchangeable insulin biosimilar, achieving rapid uptake due to payer preference and cost-effectiveness [72].
Global projections suggest that biosimilars could save healthcare systems USD 160 billion cumulatively by 2030 [73]. In India, widespread adoption could make biologics accessible to millions of patients previously unable to afford these therapies.
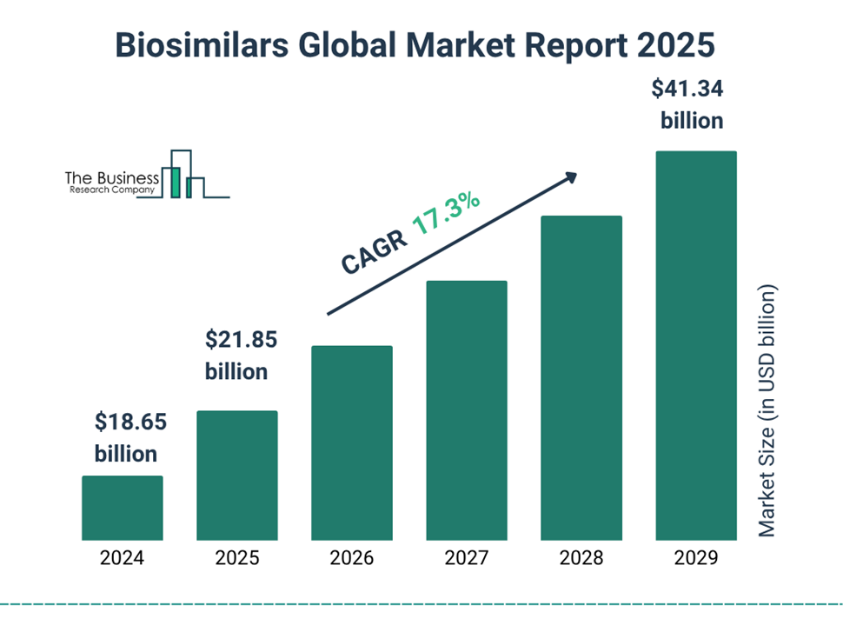
Fig. 5.6 Biosimilar Global Market Report 2025
6. Future Outlook
The biosimilar industry is entering a new era of expansion and innovation, driven by regulatory convergence, advances in analytical sciences, and global demand for affordable biologics. The period from 2025 to 2030 is expected to transform biosimilars from cost-saving alternatives into mainstream therapeutic solutions.
Next-generation analytics: High-resolution mass spectrometry, cryo-electron microscopy, and artificial intelligence (AI)-driven modeling are enhancing the precision of biosimilarity assessments [74].
Digital health integration: Real-world data (RWD) and electronic health records (EHRs) are being harnessed for post-marketing pharmacovigilance and outcome tracking [75].
Continuous bioprocessing: Adoption of single-use bioreactors and process intensification is expected to reduce manufacturing costs by 20–30%, enabling lower biosimilar prices [76].
Complex biologics: Advances in characterization may soon allow biosimilar versions of gene therapies, CAR-T therapies, and antibody-drug conjugates (ADCs), though regulatory pathways for these are still evolving [77].
Global regulatory frameworks are moving toward convergence:
This convergence will accelerate global approvals, lower development costs, and speed up patient access.
Market expansion: Biosimilars are projected to capture 60% of the biologics market by 2030, saving healthcare systems USD 240 billion globally [82].
India’s role: India is expected to become the biosimilar hub for emerging economies, leveraging its cost-effective manufacturing and regulatory reforms [83].
Healthcare equity: Biosimilars will play a central role in achieving Universal Health Coverage (UHC) goals, especially in low- and middle-income countries (LMICs) [84].
Competition risks: Policymakers must ensure that tender-based systems encourage multiple suppliers to avoid monopolies [85].
Despite promising advances, several challenges remain:
Scientific complexity: Developing biosimilars of next-gen biologics (e.g., bispecific antibodies, ADCs) will require new analytical paradigms [86].
Awareness gaps: Continued efforts are needed to educate prescribers and patients about biosimilar safety and efficacy [87].
Intellectual property barriers: Evergreening strategies by originator companies may delay biosimilar entry [88].
Global access inequities: While Europe and India embrace biosimilars, uptake in Africa and parts of Latin America remains low due to infrastructure gaps [89].
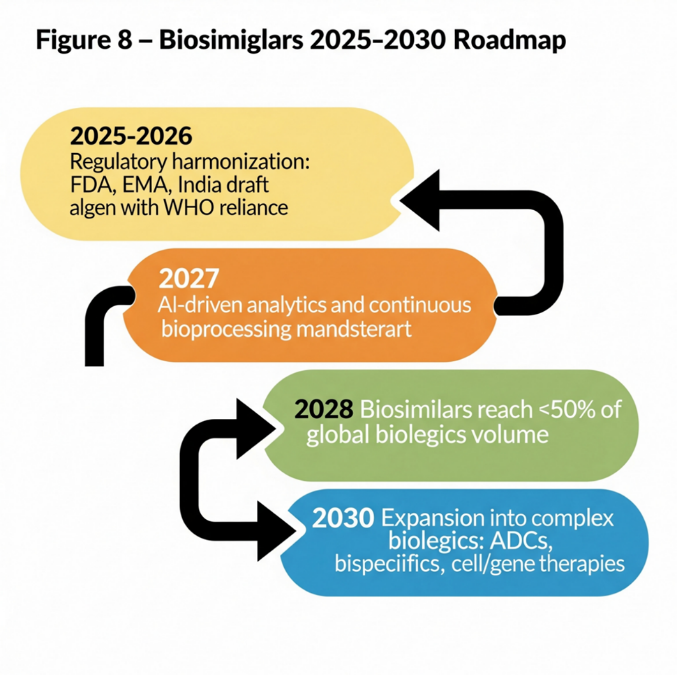
Figure 6.1: Biosimilar Roadmap 2025–2030.
Roadmap showing stages: 2025 regulatory convergence → 2027 AI-enabled analytics → 2028 global adoption 50% → 2030 expansion to complex biologics.
Partnership models: Collaborations between multinational pharma and Indian manufacturers will expand global reach.
Biobetters: Beyond biosimilars, incremental innovations may yield “biobetters” with improved delivery, stability, or dosing regimens [90].
Personalized biosimilars: Integration with pharmacogenomics could optimize therapy for patient subgroups in the future [91].
CONCLUSION
Biosimilars have moved from cautious adoption to becoming a cornerstone of sustainable healthcare systems. Evidence from randomized clinical trials, meta-analyses, and real-world registries has established their safety, efficacy, and immunogenicity profile as equivalent to originator biologics.
Regulatory agencies in the EU, US, WHO, and India are increasingly aligned, reducing duplication and fostering global trust. Market data show significant price erosion (20–70%), improved patient access, and billions of dollars in savings for healthcare systems. India, in particular, is positioned as a global biosimilar hub, supplying affordable biologics to both domestic and international markets.
Looking forward, biosimilars will play a vital role in addressing rising biologic costs, expanding access in LMICs, and meeting global health goals. With ongoing scientific advances, regulatory convergence, and increased clinician trust, the next decade will witness biosimilars reshaping the therapeutic landscape.
REFERENCES
Biological medicines, or biologics, are therapeutic products derived from living systems such as mammalian cells, yeast, or bacteria. They include monoclonal antibodies, recombinant hormones, vaccines, and therapeutic proteins, all of which have transformed modern medicine [1]. Biologics are now the backbone of therapy for cancer, autoimmune diseases, diabetes, and rare genetic disorders. However, the high cost of biologics limits their accessibility. In high-income countries, annual treatment costs for monoclonal antibodies such as trastuzumab or adalimumab range from USD 20,000 to 70,000 per patient [2]. In India, although prices are lower, they still remain unaffordable for many patients, where out-of-pocket spending accounts for nearly 60% of healthcare expenditure [3].
To address these challenges, biosimilars—biological medicines that are highly similar to approved reference products—have been developed. Unlike small-molecule generics, biosimilars cannot be exact copies due to the complex structures and manufacturing processes involved. Instead, they undergo a rigorous comparability exercise, involving detailed analytical characterization, pharmacokinetics (PK), pharmacodynamics (PD), immunogenicity, and confirmatory clinical studies [4].
Globally, the biosimilar market has grown exponentially since the first approval in Europe in 2006. As of 2025, the EMA has approved more than 90 biosimilars, the U.S. FDA has approved over 45, and India has licensed more than 120, making it a frontrunner in the developing world [5,6]. This expansion is expected to propel the global biosimilar market beyond USD 100 billion by 2030 [7].

Figure 1.1: Global biosimilar approvals by EMA, FDA, and India (2006–2025).
India has played a crucial role in biosimilar development. The Central Drugs Standard Control Organization (CDSCO) and Department of Biotechnology (DBT) issued the first biosimilar guidelines in 2012, updated in 2016, with a revised draft released in 2025 [8]. These guidelines ensure rigorous evaluation while balancing patient access. Indian companies such as Biocon, Intas, Dr. Reddy’s Laboratories, and Zydus Cadila have emerged as global players, exporting biosimilars to over 70 countries [9].
The objective of this review is to provide a comprehensive understanding of the safety, efficacy, and regulatory landscape of biosimilars in 2024–2025. The article will cover:
By integrating data, facts, figures, and real-world examples, this article seeks to highlight the transformative role biosimilars can play in addressing healthcare affordability while maintaining patient safety and clinical effectiveness.
2. Scientific Basis of Biosimilarity
The foundation of biosimilar development lies in establishing high similarity to the reference biologic without clinically meaningful differences in quality, safety, or efficacy. This principle, known as the totality-of-evidence approach, requires integration of analytical, preclinical, pharmacokinetic/pharmacodynamic (PK/PD), and clinical data [10]. The following subsections outline the scientific processes involved.
Analytical characterization is the most critical step in biosimilar development. Modern technologies allow detailed assessment of structural and functional attributes.
Table 2.1: Analytical comparability methods in biosimilar evaluation.
|
Category |
Techniques |
Purpose |
|
Primary Structure |
Peptide mapping (LC-MS/MS), Edman sequencing |
Confirms amino acid sequence identity |
|
Higher Order Structure |
Circular dichroism (CD), FTIR, NMR, X-ray crystallography |
Verifies secondary/ tertiary structure equivalence |
|
Post-Translational Modifications (PTMs) |
Glycan profiling (HPLC, LC-MS), CE-SDS, isoelectric focusing (IEF) |
Detects glycosylation, charge heterogeneity, phosphorylation, oxidation |
|
Purity & Impurities |
SEC-HPLC, CE-SDS, RP-HPLC, AUC |
Quantifies aggregates, fragments, degradation products |
|
Biological Activity |
Cell-based assays, binding affinity assays (SPR, ELISA, BLI) |
Confirms mechanism of action (MoA) and receptor binding |
|
Stability |
Stress testing (thermal, oxidative, pH, light), accelerated degradation studies |
Compares stability profile under forced conditions |
Biosimilars must demonstrate PK/PD equivalence to the reference product. Typically, healthy volunteer studies are performed to compare:
Cmax (peak concentration) and AUC (area under the curve)
Equivalence is achieved if the 90% confidence interval of the ratio of biosimilar to reference falls within 80–125% [16]. For biologics with complex dynamics, validated PD markers such as neutrophil counts (for G-CSF products) or blood glucose levels (for insulin analogues) are critical [17].
Unlike generics, biosimilars require at least one confirmatory clinical trial to assess efficacy and safety in a sensitive indication. These trials focus primarily on immunogenicity, i.e., the formation of anti-drug antibodies (ADA) and neutralizing antibodies (NAb) [18].
For example, in the PLANETRA trial (infliximab biosimilar), the incidence of ADA was comparable between the biosimilar and reference product [19].
Immunogenicity is influenced by manufacturing processes, glycosylation patterns, and even container closure systems [20].
If biosimilarity is demonstrated in one clinical indication, regulators may permit extrapolation to all approved indications of the reference biologic. This is based on scientific justification of mechanism of action, receptor binding, and pharmacology [21].
For instance, adalimumab biosimilars were initially studied in rheumatoid arthritis but subsequently approved for psoriasis, Crohn’s disease, and ulcerative colitis [22].
In India, extrapolation has been accepted for several monoclonal antibodies, provided analytical and PK/PD data support similarity [23].
Trastuzumab (Herceptin® biosimilars): Extensive analytical comparability and clinical trials in HER2-positive breast cancer confirmed biosimilarity, leading to approvals across multiple countries [24].
Insulin glargine (Semglee®): Demonstrated PK/PD equivalence and clinical efficacy, and became the first interchangeable biosimilar insulin approved by the U.S. FDA [25].
Etanercept (Erelzi® biosimilars): Confirmatory clinical trials in psoriasis supported extrapolation to multiple autoimmune conditions [26].
2.6 Significance of Scientific Evaluation
The scientific rigor behind biosimilarity ensures that patients and prescribers can trust these products to deliver the same clinical benefit as reference biologics. Importantly, over 15 years of global experience has yielded no unexpected safety concerns, reinforcing the validity of this stepwise approach [27].
3. Global Regulatory Frameworks
Biosimilars represent a regulatory challenge because, unlike generics, they are similar but not identical to their reference biologics. Regulatory authorities worldwide have therefore established science-based pathways to ensure that biosimilars meet rigorous standards of quality, safety, and efficacy. This section highlights the frameworks of the European Union (EU), the United States (US), the World Health Organization (WHO), and India, with a comparative analysis of their approaches.
The European Union (EU) was the first jurisdiction to introduce a legal framework for biosimilars in 2004, with the first approval granted in 2006 for Omnitrope® (somatropin) [28].
Guidelines: EMA established class-specific guidelines (e.g., for monoclonal antibodies, insulins, erythropoietins).
Requirements: A comprehensive comparability exercise is required, starting with analytical similarity and extending to PK/PD and confirmatory clinical trials.
Extrapolation: EMA allows extrapolation of indications if scientifically justified.
Interchangeability: In 2022, the EMA and HMA (Heads of Medicines Agencies) confirmed that all biosimilars approved in the EU are considered interchangeable with their reference product [29].
Market penetration: The EU leads globally, with biosimilars accounting for >70% of market share in epoetins and G-CSF products [30].
The US established its pathway for biosimilars under the Biologics Price Competition and Innovation Act (BPCIA) of 2010, creating the 351(k) pathway [31].
First approval: Zarxio® (filgrastim-sndz) in 2015.
Requirements: Stepwise approach, with strong emphasis on analytical similarity, animal studies (where necessary), and at least one clinical study in a sensitive indication.
Purple Book: FDA maintains a database listing all approved biologics and biosimilars, along with interchangeability status [32].
Interchangeability: Biosimilars may receive an interchangeability designation, permitting automatic substitution at the pharmacy level. Examples include Semglee® (insulin glargine), Cyltezo® (adalimumab-adbm), and Cimerli® (ranibizumab) [33].
Recent advances: In 2024, FDA proposed removing mandatory switching studies for interchangeability, signaling regulatory convergence with EMA [34].
The WHO plays a central role in shaping biosimilar regulations for low- and middle-income countries (LMICs).
First guideline: Published in 2009, introducing the concept of Similar Biotherapeutic Products (SBPs) [35].
Revision: Updated in 2022, emphasizing a risk-based, flexible approach, reducing unnecessary clinical studies when strong analytical data exist [36].
Reliance model: WHO encourages regulators in LMICs to rely on EMA/FDA-approved biosimilars to accelerate access.
Impact: Many countries in Asia, Africa, and Latin America have adopted WHO principles in their national guidelines [37].
India has emerged as a global leader in biosimilars, with over 120 approvals by 2025.
First approval: Recombinant human insulin in 2000 [38].
Guidelines:
Market: Indian companies dominate the domestic market and export biosimilars to over 70 countries. Key players include Biocon Biologics, Intas Pharmaceuticals, Dr. Reddy’s, Zydus Cadila, and Lupin [40].
Challenges: Lack of explicit interchangeability designation and limited pharmacovigilance infrastructure remain hurdles [41].
Global Regulatory Frameworks for Biosimilars
Table 3.1: Global regulatory frameworks for biosimilars.
|
Region / Regulator |
Year of First Guideline / Approval |
Key Features |
Recent Updates |
|
European Union (EMA) |
2006 (somatropin, Omnitrope®) |
First biosimilar guideline globally; stepwise comparability (quality, non-clinical, clinical) |
2022: EMA/HMA interchangeability statement |
|
United States (FDA) |
2010 (BPCIA 351(k) pathway) |
Requires analytical + PK/PD comparability; extrapolation allowed; interchangeability designation |
2024 draft: Reduces reliance on switching trials |
|
World Health Organization (WHO) |
2009 (SBP guideline) |
Provides global reference framework for member states; encourages reliance and risk-based evaluation |
2022 revision: streamlined requirements |
|
India (CDSCO/DBT) |
2012 (first guideline), 2016 (revision) |
Requires confirmatory PK/PD & clinical trials; extrapolation permitted; local bridging |
2025 draft: Accepts global reference products; aligns with WHO |
|
Japan (PMDA) |
2009 |
Similar to EMA; strong analytical focus; case-by-case clinical trial requirements |
Ongoing harmonization with ICH |
|
Canada (Health Canada) |
2010 |
Abbreviated pathway based on EMA; extrapolation permitted |
Updated guidance 2019 |
|
China (NMPA) |
2015 |
Initially restrictive, now moving toward EMA-alignment |
Expanded mAb approvals post-2020 |
|
Brazil (ANVISA) |
2010 |
Allows both standalone and comparability-based development |
Updated 2015: risk-based approach |
Key differences include:

Figure 3.1: Timeline of biosimilar regulatory milestones.
This figure illustrates how global regulatory science has evolved from the EU’s pioneering approvals in 2006 to India’s draft reforms in 2025, highlighting the trend toward harmonization.
4. Clinical Evidence on Safety & Efficacy
The acceptance of biosimilars in clinical practice depends largely on robust evidence demonstrating comparable safety, efficacy, and immunogenicity with their reference biologics. Regulators and clinicians require data from switching trials, meta-analyses, and real-world studies to establish confidence in biosimilar use. Over the last decade, and particularly in the years 2024–2025, this evidence base has expanded substantially.
Switching trials evaluate the outcomes when patients are transitioned from a reference biologic to its biosimilar. These studies focus on efficacy, safety, and immunogenicity after single or multiple switches.
NOR-SWITCH Trial (2017): A randomized trial in Norway involving 482 patients across six indications (rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and chronic plaque psoriasis). The study showed no significant difference in disease worsening between infliximab biosimilar and the originator [42].
EGALITY Trial (2017): A study of 531 psoriasis patients switching multiple times between reference etanercept and its biosimilar. Results confirmed equivalent efficacy, safety, and immunogenicity [43].
VOLTAIRE-X Trial (2021): A phase III study of 238 patients with plaque psoriasis demonstrated that switching between Humira® (adalimumab) and Cyltezo® biosimilar had no impact on efficacy or safety. This trial supported the FDA’s interchangeability designation for Cyltezo® [44].
PLANETRA and PLANETAS Trials: Evaluated infliximab biosimilars in rheumatoid arthritis and ankylosing spondylitis, showing comparable outcomes with reference products [45].
Figure 4.1: Summary of switching trial outcomes.
|
Trial |
Molecule |
Indication(s) |
Patients (n) |
Design |
Outcome |
|
NOR-SWITCH |
Infliximab (CT-P13) |
RA, AS, IBD, PsA, UC, CD |
482 |
Randomized, double-blind switch |
No difference in disease worsening |
|
EGALITY |
Etanercept (SB4) |
Psoriasis |
531 |
Multiple switches |
PASI75, ADA rates equivalent |
|
VOLTAIRE-X |
Adalimumab (BI 695501) |
Psoriasis |
238 |
Multiple switches |
Equivalent efficacy, immunogenicity |
|
PLANETRA/ PLANETAS |
Infliximab (CT-P13) |
RA, AS |
1200+ |
Double-blind switch trials |
Confirmed equivalence |
4.2 Meta-Analyses
Systematic reviews and meta-analyses consolidate data across multiple studies, strengthening the evidence base for biosimilars.
A 2020 meta-analysis covering 90 clinical trials and 14,000 patients concluded that biosimilars had no increased risk of immunogenicity or adverse events compared to reference biologics [46].
Another pooled analysis of switching studies showed no difference in efficacy endpoints (ACR20, PASI75, DAS28 remission rates) after switching [47].
A Cochrane systematic review in 2023 reiterated the clinical equivalence of biosimilars in rheumatology and oncology, supporting broader adoption [48].
Beyond clinical trials, real-world data (RWD) from registries and post-marketing surveillance provide critical insights into long-term safety and effectiveness.
DANBIO Registry (Denmark): Nationwide switch of patients from innovator infliximab to biosimilar infliximab showed stable remission rates and no safety concerns [49].
Swedish Rheumatology Quality Register: Demonstrated comparable persistence rates between etanercept biosimilars and reference products [50].
Indian Oncology Centers: Real-world studies on trastuzumab biosimilars in HER2-positive breast cancer revealed equivalent progression-free and overall survival rates [51].
Pharmacovigilance reports: The European Medicines Agency’s EudraVigilance database shows no new safety signals across 90+ approved biosimilars [52].
Immunogenicity is a major clinical consideration for biosimilars. Anti-drug antibodies (ADA) can reduce efficacy or cause hypersensitivity. However, accumulated evidence indicates comparable immunogenicity rates between biosimilars and reference biologics.
The NOR-SWITCH and EGALITY trials reported no meaningful increase in ADA incidence after switching [53].
Indian clinical studies of rituximab and trastuzumab biosimilars confirmed low and clinically irrelevant ADA rates [54].
4.5 Confidence Building in Clinical Practice
The consistent findings from randomized trials, meta-analyses, and real-world studies have reassured clinicians and regulators that biosimilars can be safely and effectively substituted for reference biologics. However, physician education and patient communication remain essential to increase trust and uptake [55].
5. Market Dynamics & Adoption Trends
The market adoption of biosimilars has been one of the most transformative developments in global healthcare economics. With patent expirations of blockbuster biologics such as adalimumab, trastuzumab, and etanercept, biosimilars have entered the market with the potential to reduce costs, increase patient access, and stimulate competition. This section examines global trends, India-specific developments, price erosion, and barriers to adoption.
Since the first biosimilar approval in 2006, the global market has grown at an annual rate exceeding 25%. According to IQVIA reports, biosimilars generated savings of over €10 billion annually in the European Union alone by 2023 [56].
Europe (EU): Europe remains the largest biosimilar market, with more than 90 approvals by EMA. Market share for certain classes is impressive: epoetins (80%), G-CSF (85%), and infliximab (70%) [57].
United States (US): The U.S. market was initially slow due to regulatory complexity and patent litigation. However, since 2019, biosimilars have gained momentum. By 2025, over 45 products are approved, and adalimumab biosimilars alone captured ~30% of U.S. market share within two years of launch [58].
Asia-Pacific: Countries like South Korea and China have rapidly advanced biosimilar production, focusing on domestic affordability and global exports [59].

Figure 5.1: Biosimilar market share in EU vs US vs India (2015–2025).
5.2 India-Specific Market
India has become a pioneer in biosimilar development among low- and middle-income countries.
Approvals: By 2025, India has approved 120+ biosimilars, the largest number outside the EU [60].
Market size: Valued at approximately INR 4200 crores (USD 5 billion) in 2025, with a CAGR of 15–18% [61].
Export potential: Indian biosimilars are exported to over 70 countries, including regulated markets such as the EU and the U.S. (through partnerships and co-developments) [62].
Leading companies: Biocon Biologics, Intas Pharmaceuticals, Dr. Reddy’s Laboratories, Zydus Cadila, and Lupin are among the top developers.
Adoption drivers: India’s cost-sensitive market encourages rapid biosimilar uptake, especially in oncology and endocrinology.

Figure 5.2: India-specific biosimilar adoption trends (approvals and sales revenue, 2010–2025).
5.3 Price Erosion
Biosimilars drive significant price erosion, improving affordability.
In the EU, tender-based procurement has resulted in 50–70% price reductions for epoetins, G-CSF, and monoclonal antibodies [63].
In the US, price erosion averages 20–35%, though competition for adalimumab biosimilars is pushing discounts closer to 50% [64].
In India, trastuzumab and insulin glargine biosimilars are priced 30–40% lower than reference products, expanding access for middle-class and rural populations [65].

Figure 5.3: Average price erosion by therapeutic class (oncology, diabetes, autoimmune, supportive care).
5.4 Barriers to Adoption
Despite proven equivalence, biosimilars face adoption challenges:
5.5 Case Examples
Adalimumab (Humira®): In the EU, biosimilar entry reduced prices by nearly 70%. In the U.S., within two years of biosimilar entry (2023–2025), originator sales dropped sharply as multiple biosimilars entered the market [70].
Trastuzumab (Herceptin®): In India, the introduction of trastuzumab biosimilars reduced treatment costs for HER2+ breast cancer patients by 30–35%, increasing accessibility in public hospitals [71].
Insulin Glargine: Semglee® became the first FDA-approved interchangeable insulin biosimilar, achieving rapid uptake due to payer preference and cost-effectiveness [72].
Global projections suggest that biosimilars could save healthcare systems USD 160 billion cumulatively by 2030 [73]. In India, widespread adoption could make biologics accessible to millions of patients previously unable to afford these therapies.

Fig. 5.6 Biosimilar Global Market Report 2025
6. Future Outlook
The biosimilar industry is entering a new era of expansion and innovation, driven by regulatory convergence, advances in analytical sciences, and global demand for affordable biologics. The period from 2025 to 2030 is expected to transform biosimilars from cost-saving alternatives into mainstream therapeutic solutions.
Next-generation analytics: High-resolution mass spectrometry, cryo-electron microscopy, and artificial intelligence (AI)-driven modeling are enhancing the precision of biosimilarity assessments [74].
Digital health integration: Real-world data (RWD) and electronic health records (EHRs) are being harnessed for post-marketing pharmacovigilance and outcome tracking [75].
Continuous bioprocessing: Adoption of single-use bioreactors and process intensification is expected to reduce manufacturing costs by 20–30%, enabling lower biosimilar prices [76].
Complex biologics: Advances in characterization may soon allow biosimilar versions of gene therapies, CAR-T therapies, and antibody-drug conjugates (ADCs), though regulatory pathways for these are still evolving [77].
Global regulatory frameworks are moving toward convergence:
This convergence will accelerate global approvals, lower development costs, and speed up patient access.
Market expansion: Biosimilars are projected to capture 60% of the biologics market by 2030, saving healthcare systems USD 240 billion globally [82].
India’s role: India is expected to become the biosimilar hub for emerging economies, leveraging its cost-effective manufacturing and regulatory reforms [83].
Healthcare equity: Biosimilars will play a central role in achieving Universal Health Coverage (UHC) goals, especially in low- and middle-income countries (LMICs) [84].
Competition risks: Policymakers must ensure that tender-based systems encourage multiple suppliers to avoid monopolies [85].
Despite promising advances, several challenges remain:
Scientific complexity: Developing biosimilars of next-gen biologics (e.g., bispecific antibodies, ADCs) will require new analytical paradigms [86].
Awareness gaps: Continued efforts are needed to educate prescribers and patients about biosimilar safety and efficacy [87].
Intellectual property barriers: Evergreening strategies by originator companies may delay biosimilar entry [88].
Global access inequities: While Europe and India embrace biosimilars, uptake in Africa and parts of Latin America remains low due to infrastructure gaps [89].

Figure 6.1: Biosimilar Roadmap 2025–2030.
Roadmap showing stages: 2025 regulatory convergence → 2027 AI-enabled analytics → 2028 global adoption 50% → 2030 expansion to complex biologics.
Partnership models: Collaborations between multinational pharma and Indian manufacturers will expand global reach.
Biobetters: Beyond biosimilars, incremental innovations may yield “biobetters” with improved delivery, stability, or dosing regimens [90].
Personalized biosimilars: Integration with pharmacogenomics could optimize therapy for patient subgroups in the future [91].
CONCLUSION
Biosimilars have moved from cautious adoption to becoming a cornerstone of sustainable healthcare systems. Evidence from randomized clinical trials, meta-analyses, and real-world registries has established their safety, efficacy, and immunogenicity profile as equivalent to originator biologics.
Regulatory agencies in the EU, US, WHO, and India are increasingly aligned, reducing duplication and fostering global trust. Market data show significant price erosion (20–70%), improved patient access, and billions of dollars in savings for healthcare systems. India, in particular, is positioned as a global biosimilar hub, supplying affordable biologics to both domestic and international markets.
Looking forward, biosimilars will play a vital role in addressing rising biologic costs, expanding access in LMICs, and meeting global health goals. With ongoing scientific advances, regulatory convergence, and increased clinician trust, the next decade will witness biosimilars reshaping the therapeutic landscape.
REFERENCES


Jitendra Maurya, Bhavini Gharia, Navigating the Evolving Landscape of Biosimilars: Safety, Efficacy, and Regulatory Advances (2024–2025), Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 1069-1086. https://doi.org/10.5281/zenodo.17086297
 10.5281/zenodo.17086297
10.5281/zenodo.17086297