
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Mallareddy Institute of Pharmaceutical Sciences, Maisammaguda, Dhulapally
Sulopenem etzadroxil is a prodrug of the penem antibacterial sulopenem. Like other beta-lactam antibacterials, it is a time-dependent inhibitor of bacterial cell wall synthesis. Probenecid is a medication used to treat gouty arthritis, tophaceous gout, and hyperuricemia. The FDA approved sulopenem etzadroxil and probenecid (Orlynvah; Iterum Therapeutics) for the treatment of uncomplicated urinary tract infections (uUTIs) caused by Escherichia coli, Klebsiella pneumoniae, or Proteus mirabilis in adult women with limited or no alternative oral antibacterial option. HPLC is the dominant separation technique to detect, separate and quantify the drug. A number of chromatographic parameters were analyzed to optimize the method like sample pretreatment, choosing mobile phase, column, and detector selection. . Method development is a broad term. In both quantitative and qualitative analysis developing a new method either for estimation of quantity of substance or to check the presence of the required component is a necessitate.
The development of the pharmaceuticals brought a revolution in human health. These pharmaceuticals would serve their intent only if they are free from impurities and are administered in an appropriate amount. To make drugs serve their purpose various chemical and instrumental methods were developed at regular intervals, which are involved in the estimation of drugs [1]. The development of pharmaceutical analytical methods represents one of the most significant aspects of drug development. Pharmaceutical analysis plays a pivotal role in ensuring the safety, efficacy, and quality of pharmaceutical products. With the continuous advancements in pharmaceutical research and development, the demand for sophisticated analytical techniques has grown exponentially [2]
Sulopenemetzadroxil
Sulopenem is a thiopenem b-lactam antibiotic with intravenous and oral formulations being developed for the treatment of infections caused by multidrug-resistant bacteria. Sulopenem etzadroxil, the oral prodrug of intravenous sulopenem, is combined with probenecid to extend its plasma half-life (1). As seen with other b-lactams, sulopenem’s bactericidal mode of action against susceptible bacteria is due to binding to cell-wall penicillin binding proteins to block peptidoglycan cross-linking and prevent further cell wall synthesis. Sulopenem possesses potent activity against species of the Enterobacterales that encode extended-spectrum b-lactamases (ESBLs) or AmpC-type (AmpC) b-lactamases that confer resistance to third-generation cephalosporins. (Probenecid was introduced more than 60 years ago as urate-lowering therapy (ULT) for gout.This drug is a uricosuric agent that inhibits active renal reabsorption of uric acid through variousurate transporters in the proximal tubular epithelial cells. The 2012 American College of Rheumatology (ACR) gout management guidelines have highlighted the importance of achieving a target serum urate (SU) < 0.36 mmol/l (6 mg/dl) for long term effective treatment of gout.
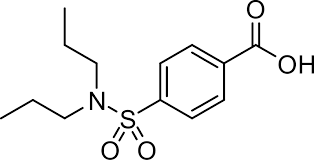
Figure 1: Structure of Probenecid
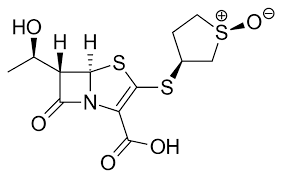
Figure 2: Sulopnemetzadroxil
Probenecid
Probenecid {4-[(dipropyl-amino) sulfonyl] benzoic acid} is a classical inhibitor of OAT and is widely prescribed for therapeutic treatment of gout and other hyperuricemia disorders. Probenecid usage for treatment of other OAT-mediated disorders such as hypertension has also been explored, along with its use to extend the plasma level of drugs identified as OAT substrates (such as -lactam antibiotics and several antiviral drugs) .In this study, probenecid was shown to reduce OAT3 mRNA and protein levels in vitro and in vivo. Administration of probenecid alone reduced influenza a virus titer in agreement with the finding that OAT3 is important for influenza a virus replication [3].
Mechanism of action of probenecid Probenecid was initially described as an agonist of theTRPV2 channels in hamster ovaries overexpressing the channel [4]. Subsequently, it was described as an agonist of TRPV2 channels in cardio myocytes with potent inotropic effects in vitro and in several in vivo animal models. The in vitro studies demonstrated that probenecid-induced agonism of TRPV2 increased intracellular calcium flux with increased CICR resulting in augmented intracellular calcium handling, particularly during diastole. Mediated Ca2+ influx, greater augmentation of SERCA activity, and increased Ca2+ release and contractility .Concurrent animal models, both healthy and diseased, confirmed the in vitro observations of increased contractility. Specifically, murine ischemic models demonstrated dramatically increased contractility after treatment with probenecid that was inversely proportional to the degree of baseline dysfunction, consistent with higher expression of myocardial TRPV2 under stress conditions [5]. Several analytical methods have been reported for Probenecid estimation like LC-MS/MS [6, 7], and UV-Visible spectroscopic method [8-9] RP-HPLC [10]
High Performance Liquid Chromatography (HPLC)
High-Performance Liquid Chromatography, also known as High-Pressure Liquid Chromatography, is a type of column chromatography that is commonly used in biochemistry and analysis to separate, identify, and quantify active chemicals. It is a popular analytical technique for separating, identifying, and quantifying each element of a mixture. HPLC is a sophisticated column liquid chromatography technology [11]. HPLC is the dominant separation technique to detect, separate and quantify the drug. A number of chromatographic parameters were analyzed to optimize the method like sample pretreatment, choosing mobile phase, column, and detector selection. The objective of this article is to review the method development, optimization and validation. HPLC method development depends on chemical structure of the molecules, synthetic route, solubility, polarity, pH and pKa value and functional groups activity etc. [12]
Principle of HPLC
The distribution of the analyte (sample) between a mobile phase (eluent) and a stationary phase is the basis for the separation principle of HPLC (packing material of the column). The structure of the analyte [13]. HPLC is a separation technique that involves: The injection of a small volume of liquid sample into a tube packed with tiny particles (3 to 5 micron (μm) in diameter called the stationary phase) where individual components of the sample are moved down the packed tube (column) with a liquid (mobile phase) forced through the column by high pressure delivered by a pump. These components are separated from one another by the column packing that involves various chemical and/or physical interactions between their molecules and the packing particles. These separated components are detected at the exit of this tube (column) by a flow-through device (detector) that measures their amount. Output from this detector is called an “HPLC” In principle, LC and HPLC work the same way except for the speed, and efficiency, sensitivity, and ease of operation of HPLC are vastly superior. Though HPLC retains major of the credits for the analytical side, the earlier one of simple Liquid Chromatography still finds applications for the preparative purpose.
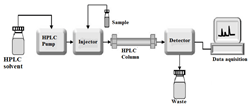
Figure 3: Component of HPLC
Types of HPLC
1. Normal Phase Chromatography (NPC)
NPC is one of the most basic modes of chromatography. This type of column consists of relatively non-polar mobile phase and relatively polar column packaging material made out of a relatively polar material with a high specific surface area, such as silica as its stationary phase.
2. Reversed Phase Chromatography (RPC)
The term reverse-phase in RPC refers to its principle which is just the inverse of the NPC; mobile phase that is relatively polar and nonpolar stationary phase. This type of column will retain non-polar compounds longer than polar compounds [14]
3. Ion exchange chromatography
In Ion exchange chromatography, retention is based on the attraction between solute ions and charged sites bound to the stationary phase. Ions of the same charge are excluded.
4. Size exclusion chromatography
Size exclusion chromatography (SEC), also called as gel permeation chromatography or gel filtration chromatography mainly separates particles based on size. It is also useful for determining the tertiary structure and quaternary structure of proteins and amino acids.
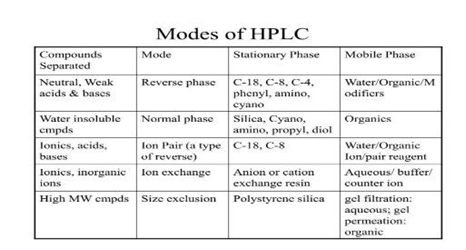
Table1: Modes of HPLC
Instrumentation of HPLC
HPLC instrumentation is a highly complex system.The combination of pumps, solvents, valves, filters,injectors, detectors, and a data-collection and processing unit makes HPLC a powerful analytical technique. The main components of an HPLC system include the solvent delivery system, the column,the detector, the recorder, and the data system. The solvent delivery system comprises a pump that can provide a steady pressure to the mobile phase. This column oven moderates the column temperature and flow meters to monitor the flow. The sample is injected into the system using various methods, such as manual injection, syringes, or autosamplers [15]
Solvent reservoir Glass or stainless steel containers capable of holding up to 1-liter mobile phase (pure organic solvents or aqueous solution of salts and buffers). Inert to a variety of aqueous and non-aqueous mobile phases. Stainless steel should be avoided for use with solvents containing halides ions solvent flow through an HPLC system begins in the solvent reservoirs, which contain the solvents used to carry the sample through the system. The solvents should be filtered through an inlet solvent filter to remove any particles that could potentially damage the system's sensitive components. Reservoirs used for storing high-performance, low-pressure liquid chromatography mobile phase. Complete reservoir system includes a plastic-coated graduated bottle, a standard cap assembly, and solvent and sparing filters.
Pump:
A pump suctions the versatile stage from the dissolvable reservoir and drives it through the framework’s column and detector. Contingent upon various components including column measurements, molecule size of the stationary stage, the stream rate and synthesis of the versatile stage, working weights of up to 42000 kPa (around 6000 psi) can be created.
Sample Injector:
The injector can be a solitary infusion or a mechanized infusion framework. An injector for a HPLC framework ought to give infusion of the liquid specimen inside the scope of 0.1–100 mL of volume with high reproducibility and under high weight (up to 4000 psi).
Column
It is the location where the separation actually occurs. The chromatographic packing material required for the separation is present in the column. Because the column hardware keeps this packing material in place, it is known as the stationary phase. It is a tube of stainless steel. 5 to 25 cm in length and 2 to 4.6 cm within. The packing material is either completely porous or only slightly porous.Stainless steel that has been cleaned typically makes up columns, which normally range in length from 50 to 300 mm and have an inside diameter between 2 and 5 mm. They typically contain a stationary phase with molecules that range in size from 3 to 10 m. Micro bore segments, or columns with inner diameters of less than 2 mm, are frequently mentioned. Ideally, during the experiment, the mobile phase and column temperatures should remain constant.
Detector:
The detector can see (detect) the individual molecules that come out (elute) from the column. High Performance Liquid Chromatography (HPLC). A detector serves to measure the amount of those molecules so that the chemist can quantitatively analyze the sample components. The detector provides an output to a recorder or computer those results in the liquid chromatogram (i.e., the graph of the detector response).
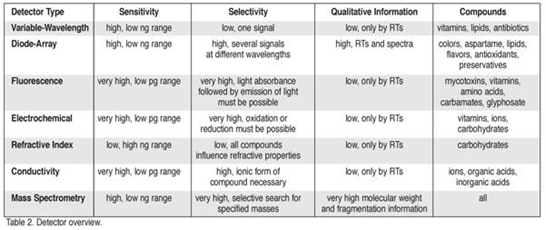
Table 2: Different detectors and types of compounds detected by them.
Data Collection Devices or Integrator
Signals from the detector might be gathered on graph recorders or electronic integrators that fluctuate in many-sided quality and in their capacity to process, store and reprocess chromatographic information. The PC coordinates the reaction of the indicator to every part and places it into a chromatograph that is anything but difficult to interpret
METHOD DEVELOPMENT AND VALIDATION ON HPLC
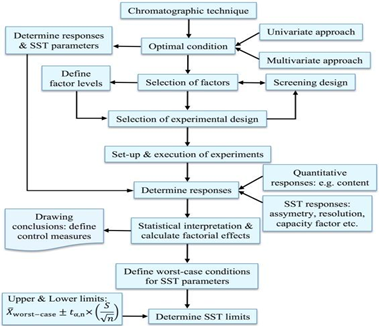
Fig 4: Flow chart of method development of HPLC
Steps involved in method development of HPLC is as follows:
1. Understanding the Physicochemical properties of drug molecule.
Physicochemical properties of a drug molecule play an important role in method development. For Method development, one has to study the physical properties like solubility, polarity, pKa and pH. pH and pKa plays an important role in HPLC method development. The pH value is defined as the negative of the logarithm to base 10 of the concentration of the hydrogen ion. pH = - log10[H3O+].
2. Selection of Chromatographic Conditions
the initial development of a method, a set of conditions, including the detector, column, and mobile phase, is chosen to generate the sample's initial "scouting" chromatograms. Commonly, reversed- phase separations using a C18 column with UV detection are employed. At this stage, the decision arises on whether to develop a gradient method or opt for an isocratic approach, each offering distinct advantages depending on the specific separation requirements and characteristics of the analytes in the sample.
3. Developing the approach of analysis
The initial chromatographic conditions for a SIM of a new entity, most important is to make sure that degradants are in solution, separated, and detected. To this effect, a diluents of 1:1 water: organic solvent is a good starting point as it will increase the likelihood of solubility of most related materials and ensure proper disintegration of solid dosage forms.
4. Sample preparations
Stability indicating method is developed by stressing the API under conditions exceeding those normally used for accelerated stability testing. Forced degradation also referred as SIMS, also can be used to provide information about degradation pathways and products that could form during storage and help facilitate formulation development, manufacturing, and packaging [16]
5. Method optimization
The experimental conditions should be optimized to get desired separations and sensitivity after getting appropriate separations. Stability indicating assay experimental conditions will be achieved through planned/systemic examination on parameters including pH (if ionic), mobile phase components and ratio, gradient, flow rate, sample amounts, Injection volume and diluents solvent type[17].
6. Method validation
During Validation of an analytical procedure is the process by which it is established, by laboratory studies, that the performance characteristics of the procedure meet the requirements for its intended use [18].
Components of method validation
The following are typical analytical performance characteristics, which may be tested during
1. System Suitability Test
This test is a requisite component of the method development, ensuring that the HPLC system and the developed method are able to provide consistent results. Injecting six replicates of EXE and GEN, the system suitability test was done, and the results were estimated through RT, theoretical plates, peak area, and tailing factor of both analytes at 256 nm. The percent relative standard deviation (RSD) of diverse parameters such as peak area, retention time (Rt), theoretical plates, and tailing factor were determined. As per US-FDA guidelines, the condition for this test is that the relative standard deviation (% RST) for RT and peak area should be less than 2%. For the tailing factor, the range should not surpass 2%, and for the theoretical plates of the column, it should be more than 2000 (N > 2000) [19]
2. Accuracy
It is the measure of how close the experimental value is to the true value. It is measured as the % of analyst recovered by assay or by spiking samples in a bling study. Accuracy should be established across the specified range (that is, line of working range) of the analytical procedure. For the assay of the drug substance, accuracy measurements are made by comparison of the results with the analysis of a standard reference material or to compare the results obtained from a second well-characterized independent procedure, the accuracy of which is stated and / or defined [20]. 3. Precision
Precision in the context of analytical methods represents the measurements made under specific conditions from several samplings of the same homogeneous material. Precision is a critical parameter for assessing the entire analytical process's reproducibility. Precision consists of two components: repeatability and intermediate precision. Repeatability is the variation experienced by a single analyst on a single instrument. It does not distinguish between variance introduced by the sample preparation procedure and that caused by the instrument or system. During validation, numerous replicates of an assay composite sample are analyzed using the analytical procedure to determine repeatability, and a recovery value is calculated. Intermediate precision refers to the fluctuation tha occurs within a laboratory on different days, with different instruments, and involving different analysts. These components of precision assessment ensure a comprehensive understanding of the reliability and reproducibility of the analytical method under varying conditions and across different operators [21]
4. Linearity and range
Linearity is the analytical method's capacity to yield test results that are exactly proportionate to the sample concentration and fall inside a given range. The relationship between the known concentrations of pure drug samples and the instrumental response was ascertained by plotting the calibration curve. A series of solutions with varying concentrations ranging from 0.006 to 100 μg/mL were created by diluting the standard stock solution with mobile phase. In the mobile phase, the nitrite calibration curve (98.00% purity) was created. To determine the calibration curve's parameters, every sample was run three times, and the data was contrived [22]
5. Detection Limit
The detection limit of an individual analytical procedure is the lowest amount of analyte in a sample, which can be detected but not necessarily quantitated as an exact value.
6. QUANTITATION LIMIT
The quantitation limit of an individual analytical procedure is the lowest amount of analyte in a sample, which can be quantitatively determined with suitable precision and accuracy. The quantitation limit is a parameter of quantitative assays for low levels of compounds in sample matrices, and is used particularly for the determination of impurities and/or degradation products [23]
7. Specificity
Selectivity and specificity are occasionally utilized conversely to depict the same idea in strategy approval. Specificity is the capacity to evaluate unequivocally the analyte near parts that may be required to be exhibit. The specificity of a test system is controlled by contrasting test results from an investigation of tests containing contaminations, debasement items, or placebo fixings with those got from an examination of tests without debasements, corruption items, or placebo fixing [24]
8. Robustness
The robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate, variations in method parameters and provides an indication of its reliability during normal usage [25-26].
REFERENCES

Krishnaphanisri Ponnekanti, Addanki Anusha, B. Raj Kamal, Anusha Polagani, Optimizing HPLC Protocols for Accurate Analysis of Sulopenem Etzadroxil and Probenecid: A Systematic Review, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 6, 4001-4010. https://doi.org/10.5281/zenodo.15731051
 10.5281/zenodo.15731051
10.5281/zenodo.15731051