1 School of Pharmaceutical Sciences, LNCT University, Bhopal, Madhya Pradesh
2 VNS College of Pharmacy
3 Arnav Institute of Pharmaceutical Sciences
4 ASPM College of Pharmacy, Sangulwadi
5 Bharati Vidyapeeth College of Pharmacy, Palus
6 Dr Bhanudas Dere College of Pharmacy, Karhe Sangamner
The scale-up of oral solid dosage forms from laboratory to pilot plant scale is a critical phase in pharmaceutical product development. Traditional scale-up approaches often rely on empirical adjustments, which may lead to variability in product quality and manufacturing inefficiencies. Quality by Design (QbD) offers a systematic, science-based, and risk-oriented framework to ensure consistent product quality during scale-up. This paper presents a comprehensive draft on the application of QbD principles in pilot plant scale-up of oral solid dosage forms, focusing on critical quality attributes (CQAs), critical material attributes (CMAs), critical process parameters (CPPs), risk assessment, design space development, and control strategies. The integration of QbD into pilot-scale operations enhances process understanding, reduces scale-up failures, and supports regulatory compliance.
Oral solid dosage forms such as tablets and capsules are the most widely used pharmaceutical formulations due to their convenience of administration, high chemical and physical stability, cost-effectiveness, and excellent patient compliance. It is estimated that more than 60–70% of marketed pharmaceutical products are available in solid oral dosage form. However, scaling up these dosage forms from laboratory-scale development to pilot plant production is a critical and complex phase in pharmaceutical manufacturing. This transition involves a substantial increase in batch size, as well as changes in equipment geometry, processing dynamics, and operational parameters, all of which can significantly impact the critical quality attributes (CQAs) of the final product.[1]
During scale-up, variations in raw material attributes (such as particle size distribution, flow properties, and polymorphic form), processing parameters (mixing time, granulation endpoint, drying conditions, compression force), and equipment characteristics (impeller speed, shear rate, die size, and compression dwell time) can lead to issues such as content uniformity failure, poor dissolution, tablet hardness variability, capping, lamination, and bioavailability inconsistency. These challenges not only affect product performance but also increase the risk of batch failure, regulatory rejection, and increased manufacturing costs.[2]

Fig. 1 Pilot Plant Scale-Up
Quality by Design (QbD), as defined by the International Council for Harmonisation (ICH) guidelines Q8 (Pharmaceutical Development), Q9 (Quality Risk Management), and Q10 (Pharmaceutical Quality System), provides a systematic and science-based approach to pharmaceutical development. QbD emphasizes a thorough understanding of the relationship between critical material attributes (CMAs), critical process parameters (CPPs), and CQAs to ensure that quality is built into the product from the early stages of development rather than being tested into the final product.[3,4]
The application of QbD principles during pilot plant scale-up enables the establishment of a Quality Target Product Profile (QTPP), identification of CQAs through risk assessment tools such as Failure Mode and Effects Analysis (FMEA) and Ishikawa diagrams, and systematic optimization using Design of Experiments (DoE). This approach facilitates the development of a design space, within which process parameters can vary without compromising product quality. Operating within the approved design space offers greater manufacturing flexibility and reduces the need for post-approval regulatory submissions.[5]
Furthermore, QbD-based scale-up supports process robustness, reproducibility, and lifecycle management, ensuring smoother technology transfer from pilot plant to commercial manufacturing. It also aligns with regulatory expectations by promoting enhanced product understanding, reduced variability, continuous improvement, and real-time quality assurance. Consequently, the integration of QbD principles in pilot plant scale-up of oral solid dosage forms significantly enhances product quality, process efficiency, and regulatory compliance, ultimately leading to safer and more effective pharmaceutical products for patients.[6]
2. PILOT PLANT SCALE-UP: AN OVERVIEW
The pilot plant serves as a critical transitional stage between laboratory-scale formulation development and full-scale commercial manufacturing. It bridges the gap by allowing systematic evaluation of formulation and process variables under conditions that closely resemble commercial production. The primary objectives of a pilot plant include:
Pilot plant scale-up typically involves batch sizes ranging from 10 to 100 times the laboratory scale, although the exact scale depends on dosage form, equipment availability, and regulatory expectations. This stage allows manufacturers to evaluate process feasibility, reproducibility, and control strategies before committing to large-scale production.[8]
For oral solid dosage forms, pilot plant operations encompass several sequential and interdependent unit operations, including blending, granulation, drying, milling, lubrication, compression, and coating. Each of these unit operations must be carefully scaled while maintaining product quality and performance.
Additionally, pilot plant studies play a crucial role in process validation readiness, equipment qualification, and risk assessment under Quality by Design (QbD) principles. Data generated at this stage supports scale-up decisions, process optimization, and regulatory compliance, ultimately reducing the risk of product failure during commercial manufacturing.[9]
3. QUALITY BY DESIGN (QBD): CONCEPT AND REGULATORY PERSPECTIVE
Quality by Design (QbD) is a systematic, science- and risk-based approach to pharmaceutical development that begins with predefined objectives and emphasizes thorough product and process understanding along with quality risk management. Unlike traditional quality approaches that rely primarily on end-product testing, QbD focuses on building quality into the product from the early stages of development.[10]
According to the International Council for Harmonisation (ICH) guideline Q8 (R2), QbD aims to ensure consistent product quality by identifying sources of variability and controlling them throughout the product lifecycle. The implementation of QbD promotes robust manufacturing processes, minimizes batch failures, and improves regulatory confidence.
From a regulatory perspective, agencies such as the US FDA, EMA, and CDSCO actively encourage the adoption of QbD principles as it enhances scientific understanding, supports regulatory flexibility, and enables continuous improvement without compromising product quality.[11]
3.1 Key Elements of Quality by Design
3.1.1 Quality Target Product Profile (QTPP)
The QTPP is a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure desired safety and efficacy. It includes dosage form, route of administration, strength, release characteristics, stability, and patient compliance considerations.[12]
3.1.2 Critical Quality Attributes (CQAs)
CQAs are physical, chemical, biological, or microbiological properties that must be controlled within predefined limits to ensure product quality. Examples for oral solid dosage forms include assay, content uniformity, dissolution rate, hardness, friability, and impurity levels.
3.1.3 Critical Material Attributes (CMAs)
CMAs refer to material-related properties of APIs and excipients that can influence CQAs. These include particle size distribution, polymorphic form, moisture content, flow properties, and bulk density. Understanding CMAs is essential for raw material selection and supplier qualification.[13]
3.1.4 Critical Process Parameters (CPPs)
CPPs are process parameters whose variability can impact CQAs and therefore must be monitored or controlled. In tablet manufacturing, examples include mixing time, granulation endpoint, drying temperature, compression force, and coating spray rate.
3.1.5 Risk Assessment
Risk assessment is a cornerstone of QbD and is performed using systematic tools such as Failure Mode and Effects Analysis (FMEA), Ishikawa (fishbone) diagrams, and Hazard Analysis and Critical Control Points (HACCP). Risk assessment helps prioritize variables and focus development efforts on high-risk factors.[14]
3.1.6 Design Space
Design space is defined as the multidimensional combination and interaction of input variables and process parameters that have been demonstrated to assure product quality. Operation within the approved design space is not considered a regulatory change, offering flexibility during manufacturing.
3.1.7 Control Strategy
The control strategy consists of planned controls derived from process understanding, including raw material controls, in-process controls, finished product specifications, and process monitoring. An effective control strategy ensures consistent performance and regulatory compliance.
3.1.8 Lifecycle Management
Lifecycle management involves continuous monitoring and improvement of the product and process throughout its commercial life. It aligns with ICH Q10 (Pharmaceutical Quality System) and supports post-approval changes based on accumulated knowledge and performance data.[15]
4. QUALITY TARGET PRODUCT PROFILE (QTPP)
The Quality Target Product Profile (QTPP) is a prospective summary of the quality characteristics that a pharmaceutical product should possess to ensure its intended safety, efficacy, and quality throughout its lifecycle. Within the Quality by Design (QbD) framework, the QTPP serves as the starting point for rational product development and provides a clear link between patient needs, clinical performance, and manufacturing considerations.[20,16]
Regulatory guidelines, particularly ICH Q8 (R2), emphasize the importance of defining the QTPP early in development to guide formulation design, process optimization, and risk assessment activities. A well-defined QTPP helps ensure that product development is patient-centric and aligned with regulatory expectations.[17]
For oral solid dosage forms, the QTPP typically includes the following key elements:
The QTPP also considers packaging requirements, administration frequency, and target patient population, particularly for special populations such as pediatric or geriatric patients.
Importantly, the QTPP acts as the foundation for identifying Critical Quality Attributes (CQAs). Each element of the QTPP is systematically translated into measurable quality attributes that must be controlled during formulation and manufacturing. Thus, the QTPP not only defines the desired end product but also guides formulation selection, process development, risk assessment, and control strategy design.[19]
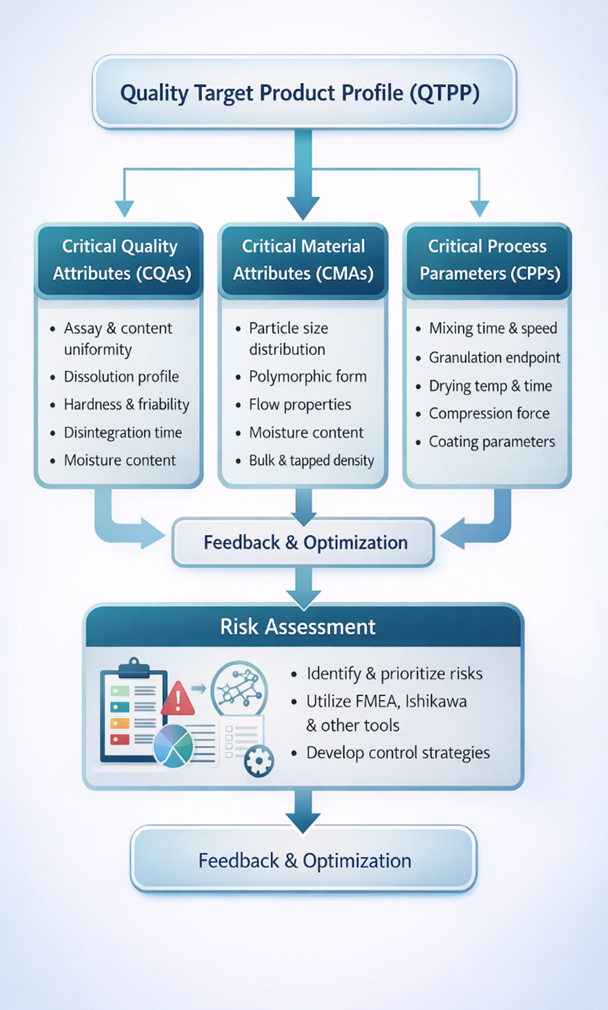
5. CRITICAL QUALITY ATTRIBUTES (CQAS)
Critical Quality Attributes (CQAs) are physical, chemical, biological, or microbiological characteristics that must be controlled within predefined limits to ensure that the pharmaceutical product consistently meets its intended quality, safety, and efficacy. According to ICH Q8 (R2), a CQA is any attribute that has a direct impact on the Quality Target Product Profile (QTPP).[21]
The identification of CQAs is a key step in the Quality by Design (QbD) approach, as it provides a scientific basis for linking formulation variables and process parameters to product performance. CQAs are established based on prior knowledge, risk assessment, and experimental studies.[22]
For oral solid dosage forms, commonly identified CQAs include:
During pilot plant scale-up, CQAs may be significantly influenced by changes in equipment size, processing time, mixing efficiency, heat and mass transfer, and environmental conditions such as temperature and humidity. Scale-dependent variations can alter granule properties, tablet density, and coating uniformity, potentially impacting final product quality.[24]
6. CRITICAL MATERIAL ATTRIBUTES (CMAS)
Critical Material Attributes (CMAs) are the physical, chemical, biological, or microbiological properties of active pharmaceutical ingredients (APIs) and excipients that have the potential to influence Critical Quality Attributes (CQAs) of the finished pharmaceutical product. Within the Quality by Design (QbD) framework, CMAs are identified through prior knowledge, experimental studies, and systematic risk assessment.[25]
According to ICH Q8 (R2), understanding and controlling CMAs is essential for ensuring consistent product quality and minimizing variability during manufacturing and scale-up.
For oral solid dosage forms, important CMAs include:
Additional CMAs may include excipient grade, surface area, porosity, and lubricant sensitivity, all of which can affect product performance and manufacturability.
During pilot plant scale-up, variability in raw material sources, supplier changes, lot-to-lot differences, and increased batch sizes can significantly influence CMAs. Such variability may lead to changes in granule characteristics, tablet mechanical strength, dissolution behavior, and stability profiles.[27]
Therefore, strict control of CMAs through material specifications, supplier qualification, incoming raw material testing, and change management systems is essential. Understanding the relationship between CMAs and CQAs enables the identification of Critical Process Parameters (CPPs) and supports the development of a robust control strategy, ensuring successful scale-up and consistent commercial manufacturing.[28]
7. CRITICAL PROCESS PARAMETERS (CPPS)
Critical Process Parameters (CPPs) are process variables whose variability has a direct and significant impact on Critical Quality Attributes (CQAs) and, therefore, must be monitored and controlled to ensure consistent product quality. As defined in ICH Q8 (R2), a CPP is a parameter that, when varied beyond a defined range, can adversely affect the quality of the finished pharmaceutical product.[29]
Identification of CPPs is achieved through process understanding, risk assessment, and experimental evaluation, particularly during pilot plant scale-up, where process behavior at increased batch sizes can be systematically studied.[30]
For oral solid dosage forms, commonly identified CPPs include:
Understanding the interrelationship between CPPs and CQAs is critical for developing a robust and scalable manufacturing process. During pilot-scale operations, Design of Experiments (DoE) is often employed to study the effects and interactions of CPPs, enabling the establishment of acceptable operating ranges and contributing to the definition of a design space.[31]
Effective control of CPPs during pilot plant scale-up ensures process consistency, reduced variability, and smooth transition to commercial-scale manufacturing, while also supporting regulatory flexibility and lifecycle management.[32]
8. RISK ASSESSMENT IN PILOT PLANT SCALE-UP
Risk assessment is a core component of Quality by Design (QbD) and plays a vital role in the successful scale-up of pharmaceutical manufacturing processes at the pilot plant level. In accordance with ICH Q9 (Quality Risk Management), risk assessment provides a systematic and science-based approach to identifying, analyzing, and controlling potential sources of variability that may impact product quality during scale-up.[33]
During pilot plant scale-up, changes in equipment size, processing conditions, material handling, and environmental factors can introduce new risks that were not evident at the laboratory scale. Therefore, structured quality risk management tools are employed to evaluate and prioritize these risks effectively.[34]
Commonly used risk assessment tools include:
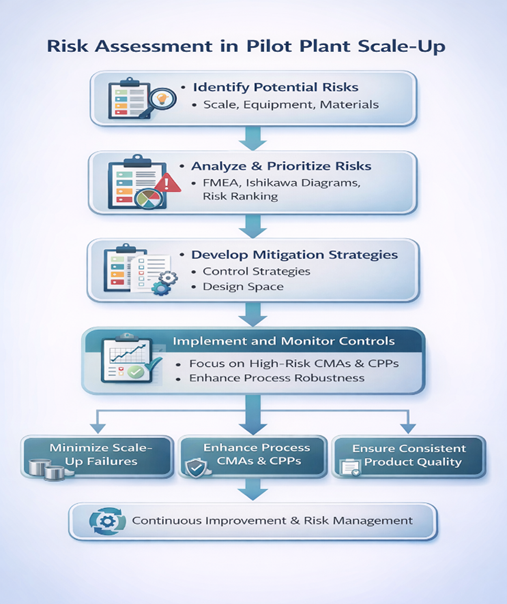
Risk assessment during pilot plant scale-up helps to:
The outcomes of risk assessment directly inform the design of Design of Experiments (DoE) studies, the establishment of and the development of an effective control strategy. Additionally, risk design space, assessment is a continuous activity that extends throughout the product lifecycle, supporting ongoing process optimization, change management, and regulatory compliance.[37]
9. DESIGN SPACE DEVELOPMENT
Design space is defined by ICH Q8 (R2) as the multidimensional combination and interaction of material attributes and process parameters that have been demonstrated to provide assurance of product quality. Operating within an approved design space is not considered a regulatory change, thereby offering significant flexibility in manufacturing operations.[38]
During pilot plant scale-up, the establishment of a design space is particularly critical, as it enables a comprehensive understanding of how Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs) interact to influence Critical Quality Attributes (CQAs) at increased batch sizes. The pilot scale provides an optimal environment to evaluate these interactions under conditions that closely resemble commercial manufacturing.[39]
The development of a robust design space during pilot plant scale-up provides several advantages, including:
Design of Experiments (DoE) is a key statistical tool used to establish and verify the design space at the pilot scale. DoE enables systematic investigation of the effects of multiple variables and their interactions on CQAs using a minimal number of experimental runs. Commonly employed DoE approaches include factorial designs, fractional factorial designs, and response surface methodologies.[41]
Data generated from pilot-scale DoE studies are used to define acceptable operating ranges, identify critical interactions, and confirm process robustness. These studies form the scientific basis for regulatory submissions and support technology transfer to commercial-scale manufacturing.
Overall, design space development during pilot plant scale-up strengthens the QbD framework by linking material attributes, process parameters, and product quality, thereby ensuring consistent performance and regulatory compliance throughout the product lifecycle.[42]
10. CONTROL STRATEGY
A control strategy is a planned set of controls, derived from comprehensive product and process understanding, that ensures the manufacturing process consistently operates within the defined design space and produces a product meeting all Critical Quality Attributes (CQAs). In line with ICH Q8 (R2) and ICH Q10 (Pharmaceutical Quality System), an effective control strategy integrates material controls, process controls, and end-product testing to maintain product quality throughout the product lifecycle.[43]
During pilot plant scale-up, the control strategy is refined and validated under conditions that closely resemble commercial manufacturing. This stage provides critical insight into process variability and helps establish controls that are both effective and practical at larger scales.
A robust control strategy typically includes the following components:
Advanced Process Analytical Technology (PAT) tools can be integrated at the pilot scale to enhance process monitoring and control. Techniques such as near-infrared (NIR) spectroscopy, Raman spectroscopy, and online particle size analysis enable real-time measurement of critical parameters, supporting real-time release testing (RTRT) and improved process understanding.[45]
11. CASE APPLICATION: QbD-BASED PILOT SCALE-UP OF TABLETS
In a typical tablet formulation, the implementation of Quality by Design (QbD) begins with the systematic definition of the Quality Target Product Profile (QTPP), which outlines the intended dosage form, strength, release characteristics, bioavailability, and stability requirements. Based on the QTPP, relevant Critical Quality Attributes (CQAs) such as assay, content uniformity, dissolution profile, hardness, friability, and disintegration time are identified.[46]
Subsequently, a risk assessment is conducted to evaluate the potential impact of Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs) on the identified CQAs. Tools such as Failure Mode and Effects Analysis (FMEA) and Ishikawa diagrams are commonly employed to prioritize high-risk variables related to raw material properties, granulation conditions, drying parameters, and compression settings.
Based on the outcomes of risk assessment, Design of Experiments (DoE) studies are performed at the pilot scale to systematically evaluate the effects and interactions of selected CMAs and CPPs. Pilot-scale batches are then manufactured using optimized process parameters identified through DoE, ensuring operation within the defined design space. These studies allow assessment of process robustness under conditions representative of commercial manufacturing.[47]
The results of QbD-based pilot scale-up typically demonstrate:
Compared to conventional trial-and-error scale-up approaches, the QbD-based strategy provides greater process predictability, enhanced regulatory confidence, and improved manufacturing efficiency. The knowledge generated during pilot-scale development supports technology transfer, process validation, and lifecycle management, ultimately leading to reliable commercial production of high-quality tablet dosage forms.[47]
12. ADVANTAGES OF QbD IN PILOT PLANT SCALE-UP
The implementation of Quality by Design (QbD) in pilot plant scale-up offers several significant advantages over conventional, empirical scale-up approaches. By integrating scientific understanding, risk-based decision-making, and systematic process optimization, QbD enhances both product quality and manufacturing efficiency.
Key advantages of applying QbD during pilot plant scale-up include:
13. CHALLENGES AND LIMITATIONS
Despite the significant advantages offered by Quality by Design (QbD) in pilot plant scale-up, its implementation is associated with several practical and operational challenges. These limitations may influence the extent and pace of QbD adoption, particularly in resource-constrained manufacturing environments.[49]
Key challenges and limitations include:
Despite these challenges, the long-term benefits of QbD—such as enhanced process robustness, reduced regulatory risk, and improved product quality—often outweigh the initial limitations. With appropriate training, phased implementation, and organizational commitment, QbD can be effectively integrated into pilot plant scale-up strategies.[50]
14. CONCLUSION
The application of Quality by Design (QbD) in the pilot plant scale-up of oral solid dosage forms provides a systematic, science-based, and risk-oriented framework for pharmaceutical development. By proactively identifying and controlling Critical Quality Attributes (CQAs), Critical Material Attributes (CMAs), and Critical Process Parameters (CPPs), QbD enables the development of robust, reproducible, and scalable manufacturing processes.
Implementation of QbD at the pilot scale enhances process understanding, reduces scale-up uncertainties, and ensures consistent product quality across different manufacturing scales. The integration of risk assessment, Design of Experiments (DoE), design space development, and control strategies facilitates a smoother and more predictable transition from laboratory-scale development to commercial manufacturing.
Furthermore, adoption of QbD principles aligns pharmaceutical development with regulatory expectations outlined in ICH Q8, Q9, and Q10, providing regulatory flexibility, supporting lifecycle management, and enabling continuous process improvement. Although QbD requires higher initial investment and technical expertise, its long-term benefits in terms of quality assurance, regulatory compliance, cost efficiency, and patient safety significantly outweigh these challenges.
In conclusion, the incorporation of QbD principles during pilot plant scale-up is not only a regulatory expectation but also an essential component of modern pharmaceutical development, ensuring the successful commercialization of high-quality oral solid dosage forms.
REFERENCES

Aniket Bisen, Amita Rai, Purvi Yadav, Sakshi Bhosale, Monika Khambalkar, Rahul Waman, Pilot Plant Scale-Up of Oral Solid Dosage Forms Using Quality by Design (QbD), Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 2796-2811. https://doi.org/10.5281/zenodo.18363271
 10.5281/zenodo.18363271
10.5281/zenodo.18363271
