
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
St. Xavier’s College, Ahmedabad (Gujarat University).
The visible light photoredox concept is rapidly used for the catalytic reactions nowadays in modern organic synthesis and it offers very efficient, selective and environmentally responsible approaches for different bond formation. This bond formation can be achieved by desulfonylation, decarboxylation, and radical-mediated cleavage to the activation of aryl halides for C–B, C–O, C–S, and related couplings. Additionally we can also use defluorinative methodologies, alkene di-functionalization, bio-orthogonal photocatalytic reactions, and visible-light-driven C–H functionalization of heteroarenes in modern organic synthesis. Through this method we not only change the functional groups and bond formation, but by this approach enables the modification of biologically active molecules , enhancing their practical relevance in pharmaceuticals and material science. This review covers the most recent progress in this field of photoredox catalysis and also include mechanistic pathway, substrate scope and its application. Remaining challenges—including catalyst design, reaction scalability, and sustainable integration—are discussed to provide future perspectives on how photoredox catalysis will continue shaping innovative strategies in synthetic chemistry.
The formation of carbon–carbon (C–C) bonds is considered one of the most fundamental and broadly applied concept in modern synthetic chemistry. Over the past few years, many different developement has been achieved in using transition-metal catalysts to activate different C–X bonds, such as C–H, C–O, and C–N, which enables the construction of different molecular compounds. Interestingly, sulfonyl-based compounds, once considered as chemically inert and highly stable, have recently been in attention as valuable precursors for C–C bond formation through visible-light-driven desulfonylation. Under mild and environmentally friendly photocatalytic conditions, sulfonyl chlorides, sulfinates, sulfonamides, and sulfones can be selectively reacted using visible light, this enlarge the scope of synthetic methods. An important advantage of these approaches is their ability to change and alter a wide range of functional groups, making it possible to modify complex, biologically active molecules at a late stage without the need for protecting groups on sensitive functionalities like hydroxyl, amine, or amide. These development highlight their important role in both large-scale synthesis and drug discovery. [1]. At the same time, the formation of aryl radicals has emerged as a powerful approach for making different types of aromatic compounds. Aryl and heteroaryl groups are commonly found in pharmaceuticals, natural products, and advanced functional materials. Traditionally, arylation reactions have been majorly depends on transition-metal catalysis, which requires expensive catalysts, air- or moisture-sensitive ligands, and harsh reaction conditions. The development in visible-light photoredox catalysis has revolutionized this area by allowing the reduction of aryl halides under mild, sustainable, and cost-effective conditions to produce highly reactive aryl radical intermediates (Figure 1). Compared with alternative precursors such as diazonium salts, iodonium salts, or triflates, aryl halides remain the most accessible and practical aryl sources. Although these compounds generally exhibit high redox potentials, advances in photocatalytic systems—including transition-metal complexes, organic dyes, and semiconductor-based catalysts—have made their reduction much more feasible. Consequently, a wide range of C–C and C–heteroatom bond-forming reactions has become possible. Importantly, these methods have also shown great utility for late-stage modification of complex bioactive molecules, opening up greater opportunities in both medicinal chemistry and materials science.[2] Alongwith organic synthesis, photoredox catalysis has also made remarkable contributions to bioorthogonal chemistry, by providing different powerful strategies to manipulate biomolecules within living systems without disturbing natural cellular processes. For a reaction to called as bioorthogonal, it must occur fastly and selectively under physiological conditions, while remaining fully compatible with aqueous environments (Figure 2). Recent development has shown that photocatalytic methods, with their mild operating conditions, tunable reactivity, and precise spatiotemporal control, are exceptionally well-suited to meet these criteria. Visible-light-driven mechanism have already been used in diverse areas, including in-situ biomolecule labeling, targeted activation, and catalytic transformations inside living cells, thereby it creates a direct connection between synthetic chemistry and biological applications. While challenges remain such as catalyst deactivation and ensuring strict orthogonality in complex biological systems photocatalysis is steadily emerging as a versatile and promising platform for expanding the toolbox of bioorthogonal reactions. These advances demonstrate the transformative impact of visible-light photoredox catalysis on C–C bond formation, aryl radical generation, and bioorthogonal chemistry. In this review, we include a summary of recent developments in these areas, with emphasis on mechanistic insights, key representative reactions, and their wide-ranging applications in organic synthesis and related methods. [3].
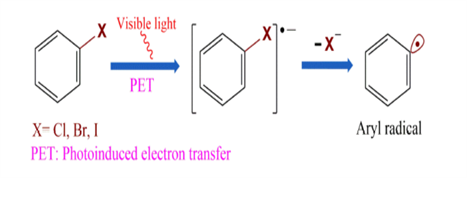
Figure 1. Generation and transformations of aryl radicals.
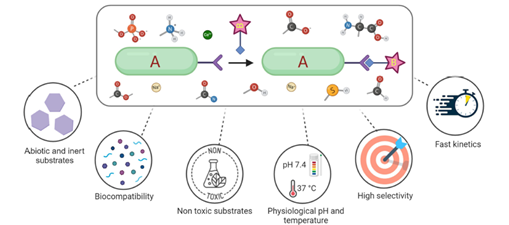
Figure 2. Overview of general reaction of bioorthogonal chemistry
2 - C–C Bond Formation under Visible Light Photoredox Catalysis
In this topic we go with the different types of transformation that could be based on the photoredox reaction and also go with the different pathways through which c-c bond formation is possible with the help of different catalyst.
2.1 Desulfonylative C–C Bond Formation
In recent years, visible-light-driven arylation and alkylation for C–C bond formation have attracted considerable interest, primarily because these reactions can proceed under mild, environmentally friendly, and often transition-metal-free conditions. Desulfonylative transformations of sulfonyl derivatives, including sulfonyl chlorides, sulfinates, sulfonamides, and sulfones, under visible-light presence have also witnessed remarkable progress. Generally, three principal mechanistic pathways are involved in desulfonylative reactions (Figure 3). In the first pathway, interaction between sulfonyl substrate 1 and the excited photocatalyst leads to the formation of sulfonyl radical anion 2 by single electron transfer (SET) process. If R² functions as a suitable leaving group, fragmentation produces the anionic species R² and sulfur dioxide, generating R¹ radical 3, which can undergo different transformations to form C–C bonds (Figure 3a). Alternatively, when R² is a non-leaving group, such as an alkyl substituent, the organic sulfonyl anion is expelled, still affording the R¹ radical 3 (Figure 3b). In a third scenario, if sulfonyl compound 1 is not directly activated by the excited photocatalyst, an external radical species 4 originating from another process—can attack compound 1. This radical substitution yields product 5 via C–C bond formation while releasing the sulfonyl radical, which is subsequently reduced to the following sulfonyl anion by interaction with another intermediate (Figure 3c).
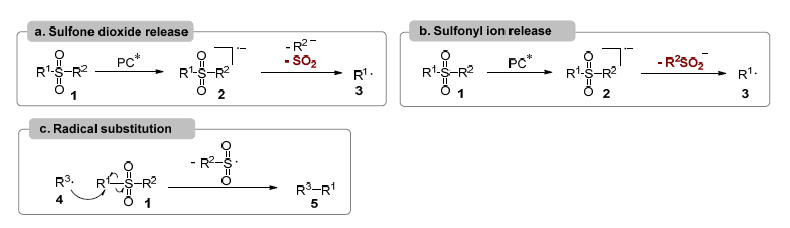
Figure 3. Mechanistic scenarios. PC* means excited photocatalyst
A. Sulfonyl Chloride: Sulfonyl chlorides are commonly used as sulfonylation reagents through the formation of sulfonyl radicals under visible-light irradiation. In the case of fluoroalkylsulfonyl chlorides, however, desulfonylation typically occurs to produce fluoroalkyl radicals, owing to their high stability. In 2011, MacMillan reported a photocatalytic method for desulfonylative trifluoromethylation of C–H bonds in a different heteroarenes (6 and 7) as well as unactivated arenes (8) (Figure 4). This mechanism enabled the selective functionalization of biologically active molecules at positions of high electron density and potential sites of metabolic vulnerability, thereby expanding opportunities for late-stage diversification.[5]
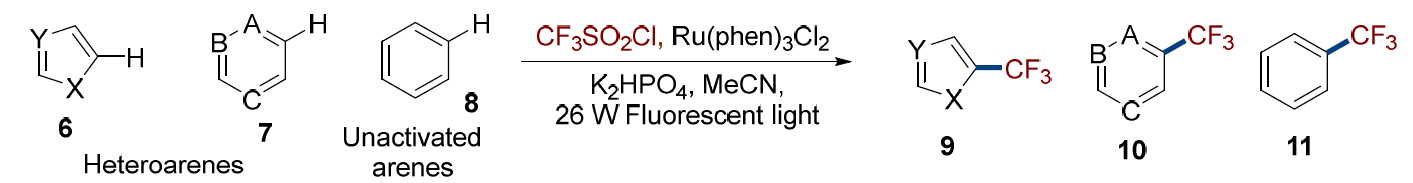
(Figure 4)
Using this method, a variety of biologically active molecules can be functionalized at positions of high electron density and metabolic sensitivity. For instance, the DNA base analogue 12, the anti-Alzheimer’s agent 13, and flavonoid 14 were regioselectively trifluoromethylated with remarkable efficiency, affording yields in the range of 85–94%.[1]

(Figure 5)
B. Sulfinates : Sulfinates, commonly considered, as sulfonylation reagents, can also undergo desulfonylation to act as precursors for alkyl radicals. Moschitto demonstrated that alkyl sulfinates undergo desulfonylative alkylation of heteroarenes when reacted under visible-light irradiation. (Figure 6) [6]
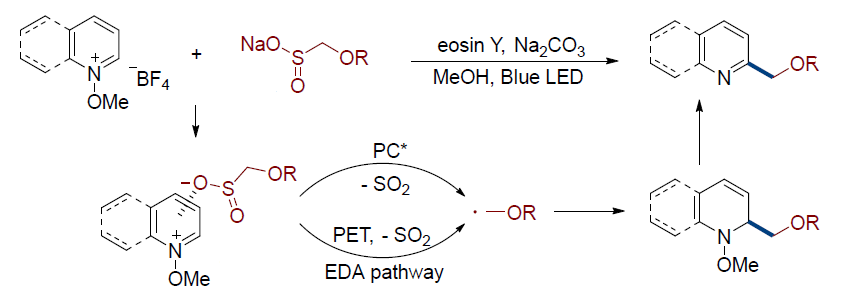
(Figure 6) hetrocycles
C. Sulphonamides: The N–S(O?) bond in sulfonamides is generally very stable, and as a result, their desulfonylative functionalization typically proceeds with a Smiles rearrangement which is initiated by radical species. In 2016, Zhang reported an efficient intramolecular aryl migration coupled with desulfonylation of sulfonamides using visible-light-induced photoredox catalysis. Based on experimental findings, a given reaction mechanism was proposed (Figure 7). In this process, the iridium photocatalyst is first excited under visible-light irradiation, followed by a single electron transfer with the sulfonamide substrate to generate a radical intermediate.[7]
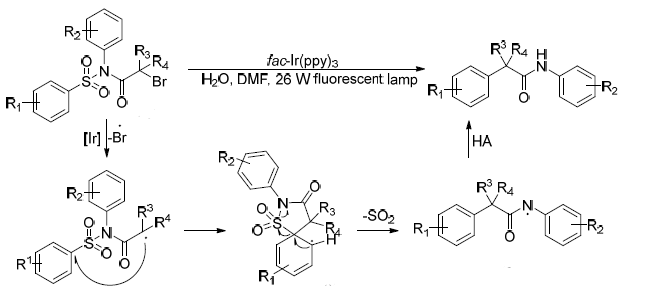
(Figure 7) Aryl sulphonamides
2.2 Decarboxylative Couplings
Carboxylic acids and their different derivatives are rampant structural motifs in both organic molecules and natural products, and their decarboxylative coupling reactions have gained significant attention. This section highlights recent progress in visible-light photoredox decarboxylative coupling, focusing on the formation of C–C as well as C–Y (Y = heteroatom) bonds.
A. Formation of C-C bond : Here, we discuss the formation of C-C bonds by using visible-light photoredox decarboxylative strategies that include decarboxylative addition, alkynylation, alkenylation, allylation and arylation.
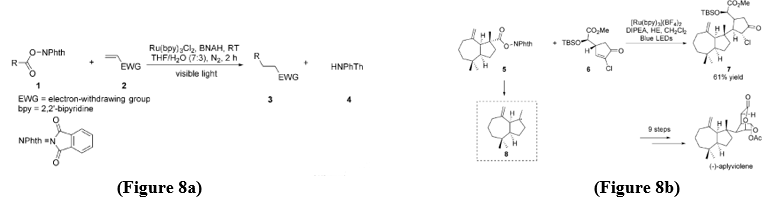
Addition Reaction - Okada, Oda, and colleagues worked on visible-light photoredox strategy for the decarboxylative Michael addition of N-(acyloxy)phthalimides 1 (Figure 8a). In this approach, electron-deficient olefins 2 act as radical acceptors, Ru(bpy)?Cl? was used as the photocatalyst, and 1-benzyl-1,4-dihydronicotinamide (BNAH) act as the reductant. The reaction proceeded in a THF–water mixture (7:3) under a nitrogen atmosphere, by this they obtained the desired product 3[9]. Overman and co-workers applied this decarboxylative methodology in the total synthesis of (-)aplyviolene. In this work, compound 7 was identified as a key intermediate and was synthesized via a visible-light-mediated decarboxylative Michael addition of N-(acyloxy)phthalimide 5 to alkene 6, using N,N-diisopropylethylamine (DIPEA) and a Hantzsch ester (HE) as reductants. This transformation proceeded through the generation of tertiary carbon radical 8 from the decarboxylation of 5 [9] (Figure 8b).
Alkynylation Reaction - Alkynes represent valuable structural motifs and versatile synthetic intermediates, and the preparation of alkyl-substituted alkynes is of significant interest. In 2015, Chen and co-workers developed a visible-light-driven reductive decarboxylative Csp³–Csp coupling method for the synthesis of aryl-, alkyl-, and silyl-substituted alkynes. (Figure 9) This transformation involved the reaction of N-(acyloxy)phthalimides with alkynyl sulfones under mild conditions, proceeding efficiently at room temperature in either organic solvents or neutral aqueous media.[10]
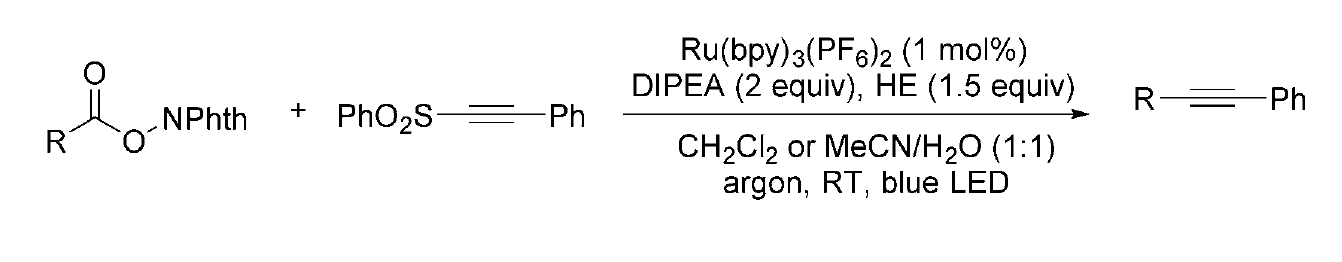
(Figure 9)
Chen and co-worker further reported a related decarboxylative ynonylation strategy employing a dual catalytic system that combined a hypervalent iodine(III) reagent with photoredox catalysis. In this method, Ru(bpy)?(PF6)2 act as the photocatalyst, α-keto acids acted as radical precursors, BI-alkyne functioned as the radical acceptor, and BI-OAc was used as an additive, enabling the efficient synthesis of ynones, ynamides, and ynoates under room-temperature conditions. (Figure 10) [11
(Figure 10)
Alkenylation Reaction – Different advances have recently been made in visible-light photoredox decarboxylative alkenylation. In 2014, MacMillan and co-workers reported a photoredox decarboxylative α-vinylation of N-Boc-α-amino acids with vinyl sulfones. In this reaction, Ir(ppy)?(dtbbpy)PF? was used as the photocatalyst, CsHCO? act as the base, and 1,4-dioxane was used as the reaction medium. The reaction was controlled at 50 °C and afforded a wide variety of allylic amines in high yields with excellent olefin geometric control. (Figure 11) [12]
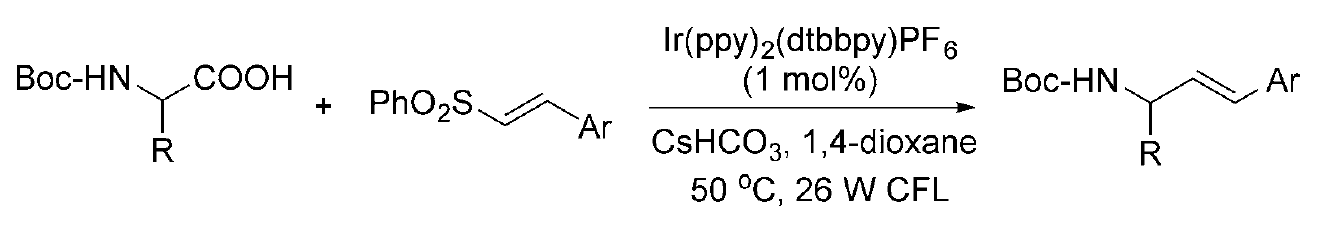
(Figure 11)
Allylation Reaction - In 2014, Tunge and co-workers developed a bi-catalytic system combining photoredox and palladium catalysis to achieve room-temperature decarboxylative allylation of amino alkanoic acids and esters. Using this method, they successfully carried out intramolecular decarboxylative allylation of phenylacetic esters and α-amino allyl esters through the cooperative catalytic action of Pd(PPh?)? and Ir(ppy)?(bpy)BF?. [13] (Figure 12a)
Arylation reaction - In 2014, MacMillan and co-workers developed a direct decarboxylative arylation of N-protected α-amino acids with electron-deficient arylnitriles using visible-light-mediated photoredox catalysis. [14] (Figure 12b)
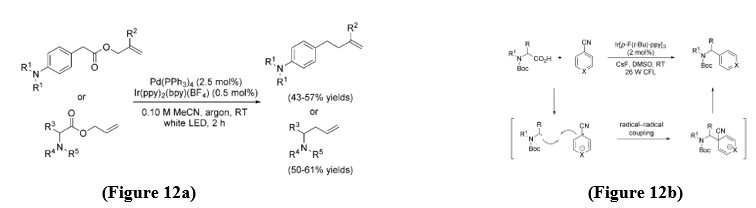
B. Formation of C-Y bonds (Y= Heteroatoms) - The visible-light photoredox decarboxylative formation of C-Y (Y=heteroatom) bonds is described and includes the formation of C-H, C-N, C-S, C-Se, and C-F bonds.
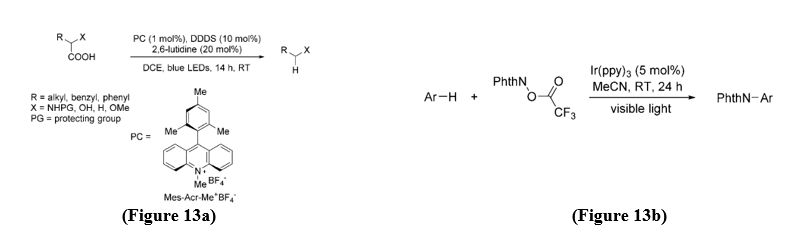
Formation of C-H bonds - Despite the significant progress made in visible-light-driven decarboxylative coupling strategies, hydrodecarboxylation of carboxylic acids has received comparatively little attention. In 2014, Wallentin and co-workers developed a decarboxylative reduction of carboxylic acids using an acridinium (Acr) photoredox catalyst (PC = Mes-Acr-Me?BF??, Mes = 2,4,6-trimethylphenyl) in combination with bis(4-chlorophenyl) disulfide (DDDS) under visible-light irradiation.[15] (Figure 13a).
Formation of C-N Bonds - Nitrogen-containing structures are abundant in natural products and pharmacologically active molecules, making the development of efficient C–N bond-forming strategies under mild conditions highly valuable. In 2014, Sanford and co-workers described a photocatalytic protocol for decarboxylative C–N amination of arene and heteroarene substrates. In this system, trifluoromethylacyloxyphthalimide served as the precursor to nitrogen-centered radical intermediates that drive the transformation.[16] (Figure13b).
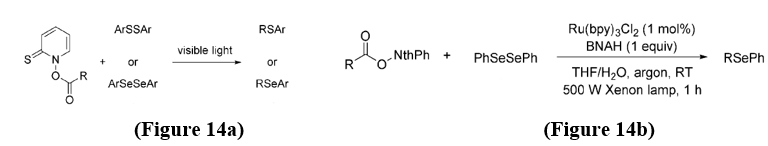
Formation of C-S and C-Se Bonds - Aryl sulfides and selenides represent important motifs in organic synthesis, materials chemistry, and drug development. Barton and co-workers developed a method of visible-light-driven decarboxylation of thiohydroxamic–carboxylic mixed anhydrides with disulfides or diselenides, this affords the corresponding sulfides or selenides in moderate to good yields. These reactions were carried out under nitrogen supply at temperatures ranging from 35 to 120 °C. [17] (Figure 14 a,b).
Formation of C-F bonds - Fluorinated molecules have great significance in both pharmaceuticals and agrochemicals, making the development of efficient fluorination strategies highly valuable. In 2014, Sammis, Paquin, and co-workers developed the first photoredox-catalyzed method for C–F bond formation. Their approach employed aryloxyacetic acid derivatives as substrates, they used Selectfluor [1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2] octane bis (tetrafluoroborate)] as the source of flourine, and Ru(bpy)?Cl? (1 mol%) as the photocatalyst, with visible-light irradiation (λ = 450 nm) driving the transformation (Figure 15a). [18].
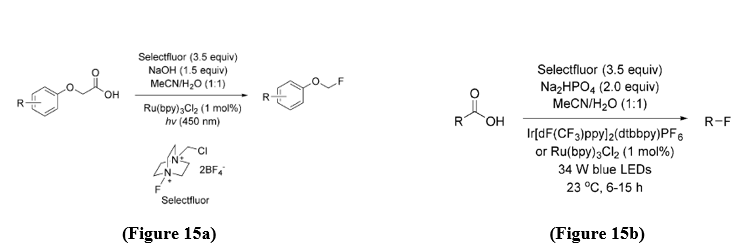
In 2015, MacMillan and co-workers reported a photoredox-catalyzed method for the decarboxylative fluorination of aliphatic carboxylic acids. This transformation was achieved using Selectfluor as the fluorine source, providing an efficient pathway for direct C–F bond formation under mild conditions (Figure 15b). [19]
2.3 Allylic Functionalization
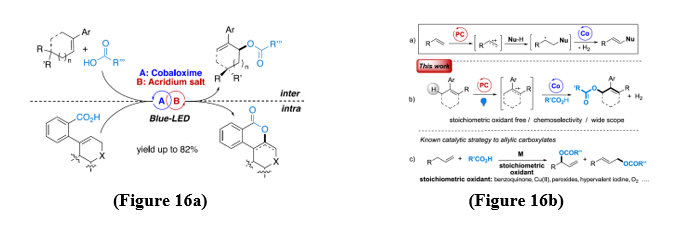
In this context, the synergistic use of Fukuzumi acridinium salts as a visible-light photoredox catalysts capable of withdraw electrons from olefins and cobalt(II/III) oximine complexes (cobaloximes) as proton acceptors has attracted considerable interest[20]. This dual system enables the direct functionalization of unactivated alkenes while simultaneously promoting catalytic hydrogen evolution. (Figure 16a,b).
2.4 Dehydroxymethylation Pathways
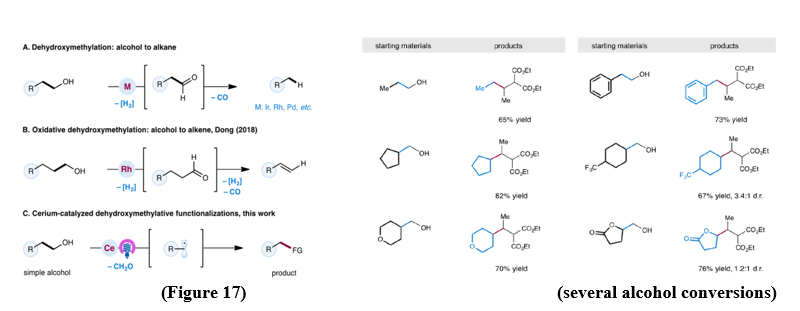
Cerium based compounds, which are both abundant and cost effective, have been used as well-organised photocatalysts due to their unique Ce(III)/Ce(IV) excitation pathways. These systems used for a wide range of radical-mediated transformations. Alcohols, although widely available and frequently used in synthesis, are rarely employed directly as carbon synthons through dehydroxymethylation, a process involving the removal of one carbon atom as formaldehyde. Conventional strategies typically depends on noble metals and external oxidants, often yielding simple alkanes . To overcome this limitation, the Dong group developed a Rhodium-catalyzed oxidative method that selectively converts alcohols into alkenes with remarkable efficiency . More recent advances in photoredox catalysis have revealed that alcohols can be convert into alkyl radicals through oxalate intermediates. Using cerium photocatalysts, a broad scope of alcohols from simple ethanol to structurally complex biomolecules can now undergo dehydroxymethylation in a mild and efficient manner. This approach opens the door to diverse cross-coupling reactions, including alkylation, amination, hydrogenation, and oxidation. [21] (Figure 17).
2.5 Alkynylation via Photoredox Catalysis
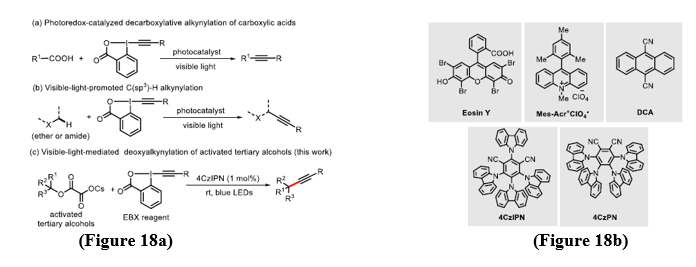
Alkynes are highly valuable material in pharmaceuticals, advanced materials, and complex molecule synthesis. Traditionally, their incorporation has depend on transition-metal-catalyzed Sonogashira coupling, which also requires elevated temperatures, specific ligands, and from side reactions such as β-hydride elimination and homocoupling. To overcome these limitations, radical alkynylation strategies using visible-light photoredox catalysis have recently gained prominence. Important advances include decarboxylative alkynylation of carboxylic acids (Figure 18a), C(sp³)–H alkynylation via benzophenone catalysis (Figure 18a), and gold-catalyzed, sunlight-mediated alkynylation of amines. Even with this progress, achieving tertiary C(sp³)–C(sp) coupling remains a significant challenge. Tertiary alcohols, which can be readily transformed into radical precursors through oxalate derivatives, gives a promising solution. Baes on the work of MacMillan and Overman, a method of direct deoxyalkynylation of tertiary alkyl cesium oxalates with EBX reagents has been developed (Figure 18a). Using 4CzIPN as a photocatalyst in dichloromethane with DMF as an additive under blue LED irradiation, this method provides high yields under mild and practical conditions. Overall, photoredox-enabled alkynylation complements classical approaches by broadening the scope of C(sp³)–C(sp) bond formation in a sustainable and operationally simple manner. [22]. During the experimental studies, a range of organic photocatalysts—including DCA, 4CzPN, Mes-Acr?ClO??, and Eosin Y(Figure 18b)were tested for their ability to give the deoxyalkynylation reaction. From these, 4CzIPN consistently gives better results than others by giving the highest yields. This performance of 4CzIPN underscores the critical role that photocatalyst structure and redox potential play in governing the efficiency of radical alkynylation. [22]
2.6 Radical-Mediated C–C Bond Cleavage and Reorganizati
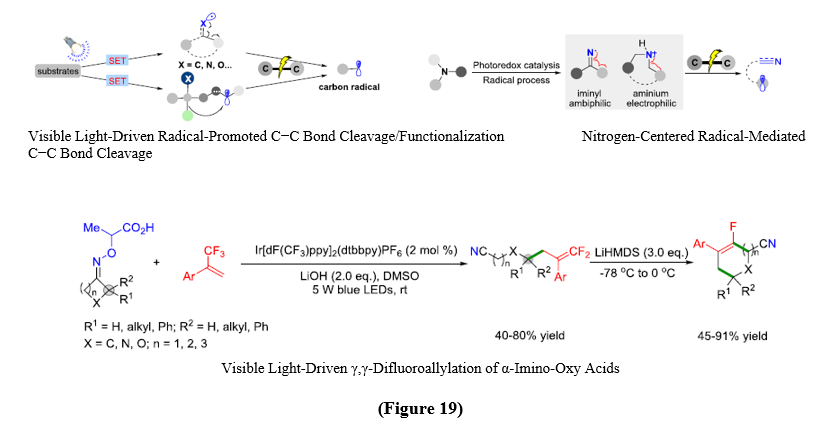
Visible-light photoredox catalysis has recently gained prominence as a sustainable alternative conventional thermal approaches for C–C bond activation, which require harsh conditions and suffered from poor selectivity. A particularly powerful strategy relies on nitrogen-centered radicals (NCRs), especially iminyl radicals, which can be generated under mild conditions from oxime esters or hydroxylamine derivatives via oxidative or reductive single-electron transfer (SET). The iminyl radicals which formed readily undergo β-C–C bond cleavage, triggering ring-opening fragmentation to produce cyanoalkyl radicals. These intermediates are highly versatile and can be captured by diverse π-systems,which enables transformations such as alkynylation, difluoroallylation, sulfonylation, and Heck-type olefinations. Among these, visible-light-driven iminyl radical Heck couplings is important, as they provide direct access to functionalized alkenes with excellent selectivity and tolerance toward a wide range of functional groups. A notable step froward in this area is the development of a dual cobaloxime–photoredox catalytic system, in which α-imino-oxy acids undergo oxidative decarboxylation to generate iminyl radicals that promote selective C–C bond clevage. The ensuing alkyl radicals are catched by cobalt, enabling radical-olefin coupling followed by β-hydride elimination to equip nitrile-substituted alkenes with excellent E-selectivity. This reaction proceeds under visible-light irradiation without any need of external oxidants, while releasing only CO?, H?, and acetone as non-hazardous by-products. This method show broad substrate tolerance, accommodating aromatic, ester, and heteroaryl functionalities, and highlights the alliance between photoredox catalysis and cobaloxime in driving radical-mediated C–C bond cleavage and reorganization. Collectively, this method provides a powerful platform for the mild and sustainable formation of functionalized alkenes and nitriles. [23] (Figure 19).
3 - C–X (Heteroatom) Bond Formation under Visible Light
3.1 From Aryl Halides Reduction of aryl halides to aryl radicals
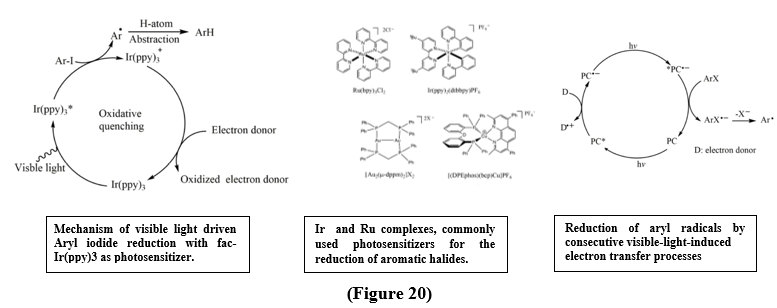
Aryl groups are very important in organic synthesis, with wide application in natural products, pharmaceuticals, and advanced materials. Old method of arylation strategies were predominantly depend on transition-metal catalysis, which, while highly effective and requires expensive catalysts, carefully tuned ligands, and harsh reaction conditions. In opposite to this, visible-light photoredox catalysis has emerged as a greener and more versatile platform, allows the mild generation of aryl radicals from inexpensive raw material. Among these, aryl halides stand available as attractive precursors due to their ready availability and low cost. Their property of high reduction potentials and the strength of the C–X(heteroatoms) bonds particularly in aryl chlorides pose formidable challenges for traditional photocatalysts, limiting the scope of such transformations. [24]. Recent developments have significantly expands the strategies available for aryl radical generation. Methods such as consecutive photoinduced electron transfer (conPET) and electron-primed photocatalysis have enabled access to reduction potentials, making the activation of inert aryl chlorides. By evading the intrinsic limitations of conventional single-electron transfer processes, these method now permit net-reductive couplings under mild conditions. As a result, a wide range of transformations including hydroarylation, borylation, and diverse C–heteroatom bond formations can be done with high efficiency. Together, these advances establish visible-light-mediated reduction of aryl halides as a strong and versatile platform, which not only overcomes long-standing challenges but also expands the synthetic C–C and C–heteroatom bond construction. [24].
C–X (B, C, O, P, S, Se) bond formation without external photocatalysts
In addition to photocatalyst-mediated reaction, an emerging concept involves photocatalyst-free methedologies, where aryl halides directly engage with visible light to undergo homolytic C–X bond cleavage. The resulting aryl radicals can be efficiently trapped by a wide range of nucleophiles or radical acceptors, this enables the construction of different C–X bonds including C–B, C–C, C–O, C–P, C–S, and C–Se Bonds . These methods shows the intrinsic photoactivity of aryl halides and reflects their versatility as radical precursors. Importantly, this type strategies minimize dependence on expensive transition-metal catalysts and external photocatalysts, providing greener, more economical, and operationally simple methods for arylation reactions. [25]
3.2 C–F Bond Activation and Defluorinative Reactions (Ar-F bond formation )
3.2.1 Synthesis of Fluorinated Aromatic Compounds – Different fluorinated aromatic compounds can be synthesized using a different types of photocatalysts. The insertion of fluoroalkyl groups into organic molecule is highly significant transformation, as these groups impart remarkable effects on molecular properties. Such insertion typically enhance lipophilicity, metabolic stability, and resistance to oxidative processes, while also increase bioavailability and biological activity. In addition to this , fluoroalkyl substituents can afford water-repellent characteristics and overall molecular robustness, making them highly efficient for pharmaceuticals, agrochemicals, and advanced materials. [26]
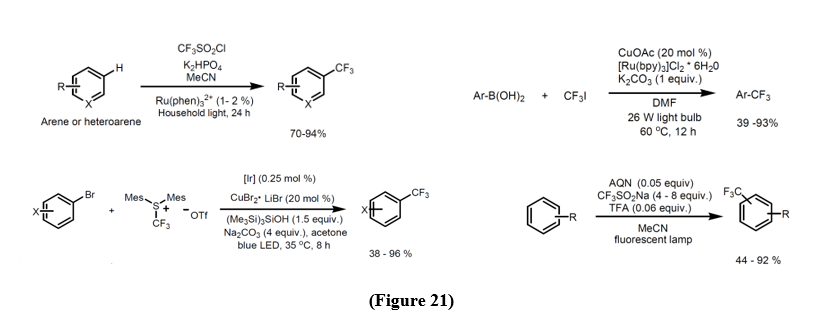
• Trifluoromethylation of Arenes - CF3SO2Cl [27], CF3I [28], Hypervalent Iodine and Sulfur Compounds [29], Trifluoromethyl Acid and Anhydride [30], Sodium Triflinate [31]. (Figure 21).
• Trifluoromethylthiolation and Trifluoromethoxylation of Arenes - The Wangelin group developed an efficient method for the radical aromatic trifluoromethylthiolation of arene-diazonium salts, which proceeds with the generation of aryl radical intermediates(Figure 22) [32]. Along with this, trifluoromethoxylation of arenes has been achieved using either radical-based fluoroalkoxylating reagents or nucleophilic trifluoromethoxylating sources. Among these methods, the photocatalytic route to trifluoromethoxyarenes is particularly efficient . Ngai and co-workers developed a catalytic intermolecular trifluoromethoxylation of (hetero)arenes under visible-light photocatalytic conditions, representing a valuable advance in accessing these highly functionalized fluorinated motifs. [33]
Figure 22)
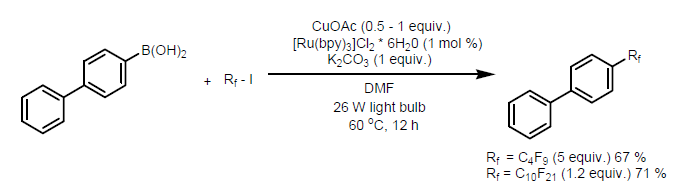
• Perfluoroalkylation of Arenes - The reaction condition developed by Ye and coworkers for trifluoromethylation of arylboronic acids using CF3I in a Cu/Ru dual catalytic system can also be used for perfluoroalkylation reactions. Using commercially available perfluorobutyl and perfluorodecyl iodides, the respective perfluoroalkylated biphenyls were obtained in good yields. (Figure 23) [34]
(Figure 23)
• Monofluorination of Arenes - The concept of visible-light-driven photocatalysis has been proved efficient for the direct C-F bond formation. Fukuzumi and coworkers reported the photocatalytic method using tetraethylammonium fluoride tetrahydrogen fluoride salt (TEAF•4HF) as fluorinating reagent in the presence of 3-cyano-1-methylquinolinium perchlorate (QuCN+ClO4−) as photocatalyst. (Figure 24) [35]
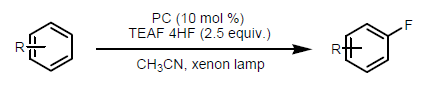
(Figure 24)
3.2.2 Radical pathways for C–F cleavage (visible-light photoredox) – Photocatalytic reduction gives an effective method for C–F bond cleavage. Several catalysts have been used for this transformation, including Ir[dF(CF?)ppy]?(dtbbpy)PF?, Ir(ppy)?(dtbbpy)PF?, 4CzIPN, 4DPAIPN, fac-Ir(ppy)?, Rose Bengal (RB), Eosin Y, and Ru(bpy)?Cl?. [36]
• Polyfluorinated Arenes - The Weaver group shown the replacement of fluorine atoms in perfluorinated arenes with hydrogen, alkyl, alkenyl, or electron-rich aryl groups via photocatalytic C–F bond cleavage. Under visible-light irradiation, the excited fac-Ir(ppy)?* is single-electron reduced by iPr?NEt (DIPEA) to generate the stronger reductant fac-Ir(ppy)??. This species reduces perfluorinated arenes to form perfluoroaryl radical anions, which then force out fluoride to give perfluoroaryl radicals. The resulting radicals can abstract a hydrogen atom from the amine radical cation, yielding the hydrodefluorination (HDF) product. (Figure 25a,b) [37].
• gem-Difluoroalkenes - The sp²-hybridized C–F bonds of gem-difluoroalkenes can undergo visible-light-mediated reaction via either radical–radical cross-coupling or an addition–elimination pathway. In 2016, the Hashmi group reported the first method of visible-light-induced defluorinative monofluoroalkenylation of tertiary amines with gem-difluoroalkenes, which proceeds through a radical–radical coupling mechanism. (Figure 26) [38]
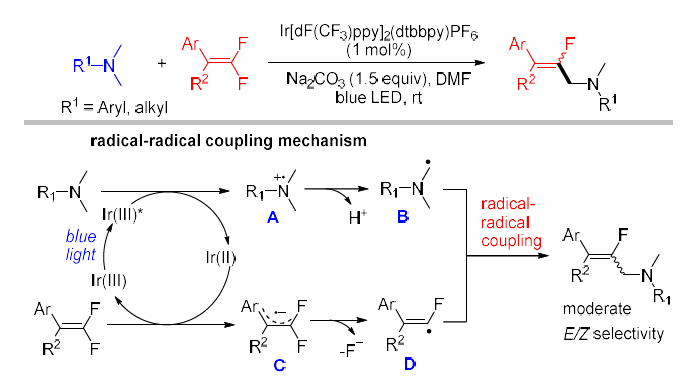
(Figure 26)
•Trifluoromethyl Arene- Gouverneurdeveloped the hydrodefluorination of trifluoromethylarenes bearing electron-withdrawing groups (EWGs) using 4-DPAIPN as the photocatalyst and 4-hydroxythiophenol (4-HTP) as the hydrogen atom donor. (Figure 27) [39]

(Figure 27)
• Trifluoromethyl Alkenes The C-F bond cleavage of trifluoromethyl alkenes usually proceeds through the radical addition and fluoride elimination pathway. Alkenes are good radical acceptors while the attachment of a CF3 group further lowers the LOMO of the C-C double bond, making radical addition to alkenes easier. To access the alpha-CF3 carbon ion B necessary for fluoride elimination, the excited photocatalyst (PC*) must be reductively quenched by a radical precursor to give PC− and radical R•. The addition of R• to alpha-CF3 alkenes provide alpha-CF3 alkyl radical A, which is SET reduced by PC−, affording anionic intermediate B concomitant with the regeneration of ground-state photocatalyst (PC). Finally, beta-fluoride elimination from B leads to gem-difluoroalkene products. (Figure 28) [40]
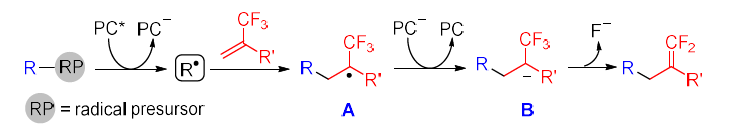
(Figure 28)
3.3 Thiol–ene and Thiol–yne Reactions
Thioethers are the significant class of organosulfur compounds that is present widely in nature and medicine. Drugs like Montelukast (asthma therapy), Butoconazole (antifungal), and Ranitidine (anti-ulcer) shows the importance of the C–S bond in pharmaceuticals. In synthetic chemistry, thioethers are also valuable intermediates for preparing sulfoxides and sulfones, making them essential building blocks in material science. A particularly effective method for their synthesis is the thiol–ene/yne (hydrothiolation) reaction, used for its simplicity and atom economy. Conventional radical-based methods were depends on thermal or UV activation, which have poor selectivity and side reactions. To overcome these limitations , visible-light photoredox catalysis has used as a sustainable and mild strategy. Since early contributions by Yoon and Stephenson, this method has expanded rapidly, providing selective and efficient routes to C–S bond formation.[41]. This part summarize developments of different methods in visible-light photoredox-catalyzed thiol–ene/yne reactions ,with emphasis on anti-Markovnikov and Markovnikov selectivity in synthetic methodologies.
3.3.1 Photoredox Catalytic Anti-Markovnikov-Selective Thiol-Ene/Yne Reactions
Due to the high reactivity of sulfur, thiol–ene/yne reactions generally follow a radical pathway, producing anti-Markovnikov products under visible-light photoredox catalysis. Since the initial works of Yoon and Stephenson, diverse systems have been designed to access structurally varied organosulfur compounds.
• Photocatalytic Anti-Markovnikov Thiol-Ene Reactions for Alkyl Thioethers
In 2015, Greaney’s group developed a visible-light-driven thiol–ene reaction using TiO? as a recyclable and inexpensive photocatalyst, it shows that inorganic semiconductors can replace transition-metal catalysts. The mechanism involved thiyl radical generation through photoinduced electron transfer, leading to alkyl sulfides via anti-Markovnikov addition(Figure 29a) . In the same year, Fadeyi’s group developed as method in which Bi?O? used as photocatalyst, capable of late-stage functionalization of complex peptides. The mechanism form BrCCl?-derived radicals to mediate thiyl radical formation, enabling efficient synthesis of alkyl thioethers with anti-Markovnikov selectivity . (Figure 29b) [42].

• Photocatalytic Anti-Markovnikov Thiol-Yne Reactions for Alkenyl Thioethers
As compared to thiol–ene chemistry, thiol–yne reactions are less developed. In 2016, Ananikov’s group develop a method in which they introduced the first metal-free, visible-light thiol–yne reaction using Eosin Y as an organophotocatalyst. This reaction, enhanced by a 3D-printed photoreactor, enabled cost-effective synthesis of alkenyl sulfides with excellent anti-Markovnikov selectivity(Figure 30). The high regioselectivity was rationalized by DFT studies, which indicated that the secondary radical formed via anti-Markovnikov addition was more stable. [43]

(Figure 30)
• Photocatalytic Anti-Markovnikov Thiol-Ene/Yne Reactions for β-Hydroxysulfides/β-Keto Sulfides
The insertion of additional functional groups during hydrothiolation enables the one-step synthesis of complex thioethers. Shah’s group (2019) showed that alkenyl/alkyl radicals generated in thiol–ene/yne reactions could undergo oxygen insertion, cycloaddition, or nucleophilic addition, which produce α,α-aminothio-substituted carbonyl compounds under mild conditions. These products, bearing weak C–S bonds, allowed for further selective functionalization, thus expanding synthetic utility. (Figure 31) [44]
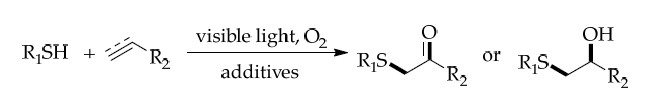
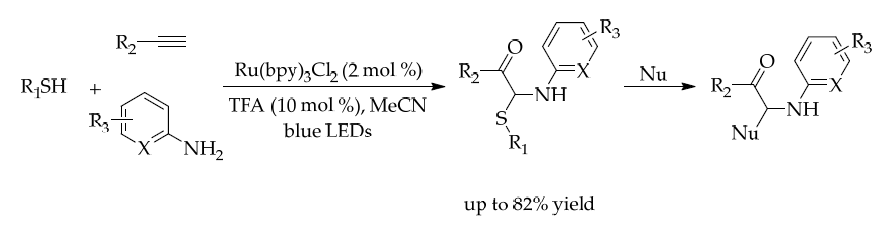
(Figure 31)
• Photocatalytic Anti-Markovnikov Thiol-Ene/Yne Reactions for Sulfoxides
Sulfoxides are basically prepared by oxidation of thioethers, but traditional methods require excess oxidants and risk overoxidation to sulfones. Photoredox catalysis has gives a greener alternative : Wang (2017) used visible light and air as oxidant to achieve selective sulfoxide synthesis from thiols and alkenes[45] , while Aleman used Eosin Y with atmospheric oxygen to obtain different sulfoxides[46] . To overcome the limitation of unrecyclable catalysts used in ang ang Aleman work, Singh developed a reusable photocatalyst, NHPI, suitable for tandem thiol–ene/oxidation reactions. The mechanism involved thiyl radical formation, anti-Markovnikov alkyl radical addition, and subsequent oxidation to sulfoxides. (Figure 32) [47].
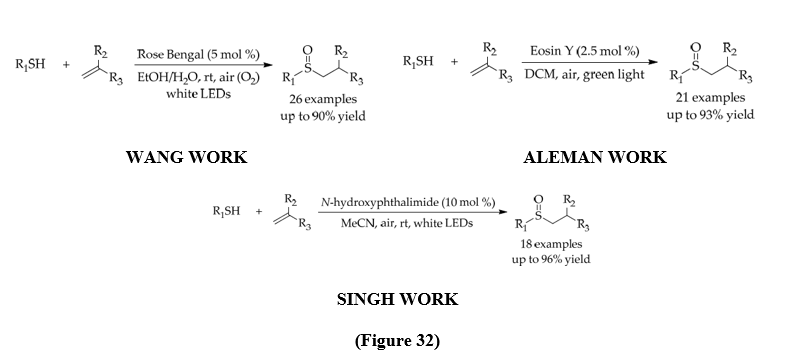
3.3.2 Photoredox Catalytic Markovnikov-Selective Thiol-Ene/Yne Reactions
Unlike the well-developed anti-Markovnikov pathway, achieving Markovnikov-selective hydrothiolation under visible light remains challenging. The difficulty arises mainly due to Kharasch effect and catalyst becomes poisonous by sulfur species, due to this efficient catalytic systems cannot be developed. Rather than this, a few successful strategies have been developed in recent years.[48]
• Photocatalytic Markovnikov Thiol-Ene Reactions for Branched Alkyl Thioethers
In 2020, Lim’s group developed a visible-light photoredox method for hydrothiolation of enamides and enecarbamates, giving N,S-acetal products with high Markovnikov selectivity. The mechanism involved thiolate anion formation, hydrogen addition at the β-position, radical oxidation to a carbocation, and trapping by thiolate to yield the branched products. (Figure33a) Later, a photoredox/cobalt dual catalytic system also developed which enables Markovnikov hydrothiolation of styrenes with thiols. The reaction utilized a protected amine directing group for cobalt coordination, allowing efficient C–S bond formation with broad substrate scope and functional group insertion. This system also used for gram-scale synthesis, showing its important practical value. (Figure 33b) [49].
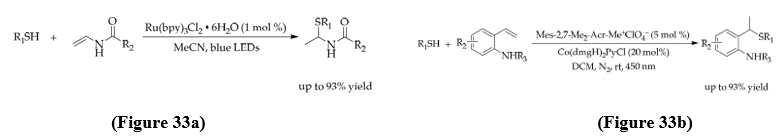
• Photocatalytic Markovnikov Thiol-Yne Reactions for Branched Alkenyl Thioethers
In 2017, Lei’s group developed a visible-light radical addition of aryl sulfonic acids to terminal alkynes, producing α-substituted vinyl sulfones with high Markovnikov selectivity. For vinyl sulfides it have moderate yields, and the mechanism involved a radical cross-coupling pathway. (Figure34a). Subsequently, Ananikov’s group developed a metal-free photoredox system that delivered four types of vinyl sulfides, including Markovnikov products, from thiophenols and alkynes. By studying the mechanism using ESI-MS and DFT calculations suggested a pathway involving thiyl radical addition, alkenyl radical stabilization, and elimination to give Markovnikov-type sulfides. (Figure 34b) [50].
.4 Remote C–H Functionalization (Borylation and Heteroaromatic Systems)
Alkyl boronic esters are important intermediates in organic synthesis because they can undergo different functional group transformations. Traditional methods for their preparation involve multistep procedures and prefunctionalization, which have lower efficiency. In opposite to this, direct C(sp³)–H borylation is an atom-economical and orthogonal route. Different advances happen in C(sp²)–H borylation, but achieving selective C(sp³)–H borylation, specially at sterically hindered tertiary sites, remains challenging due to limitations such as harsh reaction conditions, reliance on directing groups, and catalyst deactivation.[51]. On the other hand, imidazo[1,2-a]pyridines is an important class of nitrogen-containing heterocycles widely present in pharmaceuticals, natural products, and functional materials. They have a broad spectrum of biological activities, and several clinically used drugs are derived from this heterocycle. Functionalization of these hetrocycle through C–H bond activation gives a direct and easy way to modify structures, but existing methods often have limitations such as the use of strong oxidants, expensive catalysts, and limited sustainability. Recently, photochemical methods have emerged as a greener and more versatile alternative, enabling selective C–H functionalization via single-electron transfer (SET) pathways under mild conditions.[52]
3.4.1 Metal-free C(sp³)–H borylation strategies
Transition-metal-catalyzed C–H borylation is the most common method, and significant progress has been achieved with different transition metals such as Ir, Rh, Co, Ru, and Pd. A less used approach is the borylation of carbon-based radicals generated by hydrogen atom transfer (HAT). In 2020, the Aggarwal group employed a Cl°− assisted HAT strategy to achieve the primary-selective C(sp3)–H borylation of alkanes and silanes in the presence of B2cat2 and alkoxyphthalimide under mild conditions(Figure 35) . Later, Xia, Dai, and Aggarwal groups developed an iron/copper-catalyzed C(sp3)–H borylation through photoinduced ligand-to-metal charge transfer (LMCT). Gevorgyan group also developed a metal-free radical a-C–H borylation of aliphatic amines.
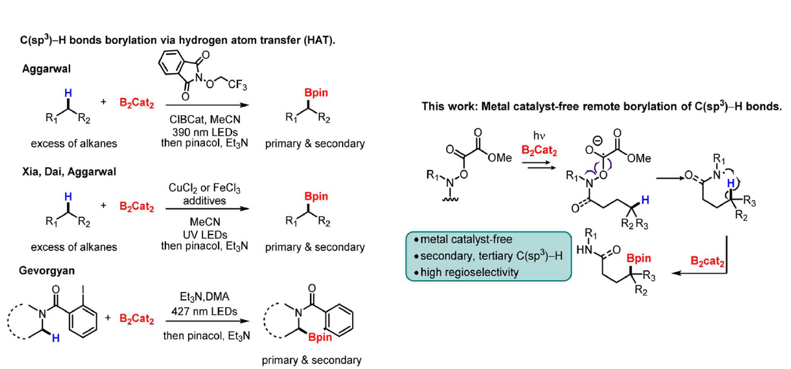
(Figure 35)
3.4.2 Direct C–H functionalization of imidazo[1,2-a] pyridines
• C3-Fluoroalkylation of Imidazo[1,2-a] pyridines
Fluoroalkyl groups are widely used in pharmaceuticals and materials science because they are able to alter the chemical and biological behavior of heterocycles. Among these, imidazo[1,2-a]pyridine derivatives have attracted attention, and several strategies have been developed for their fluoroalkylation. A common reagent for introducing a CF? unit is Langlois’ salt (CF?SO?Na). In 2016, Rueping and co-workers developed a method for C3-trifluoromethylation of imidazo[1,2-a]pyridines under near-UV light (350 nm) using 4,4′-dimethoxybenzophenone as a photocatalyst, yield was modest (42%).(Figure 36) [53]
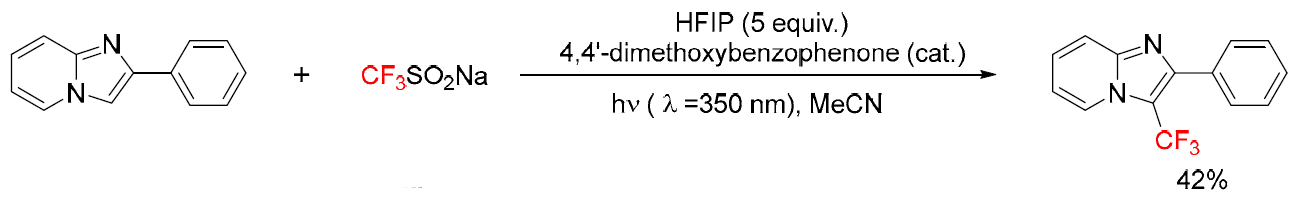
(Figure 36)
• C3-Cyanomethylation of Imidazo[1,2-a]pyridines
In organic and drug synthesis, cyano groups are important intermediates since they can be transformed into different functional groups. Sun and co-workers (2017) reported a visible light-driven mechanism for C3-cyanomethylation of imidazo[1,2-a]pyridines using bromoacetonitrile and fac-Ir(ppy)? as photocatalyst. The reaction allows both electron-donating and electron-withdrawing substituents, giving products in moderate to excellent yields (12–96%). Substrates with aryl or alkyl groups at the C2 position also reacted well, and a photocatalytic mechanism was proposed. (Figure 37) [54]
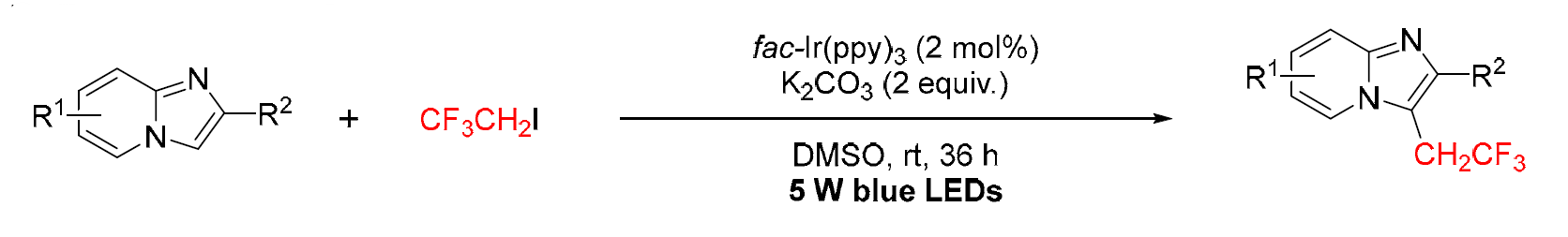
(Figure 37)
4 - Applications in Bioorthogonal and Biological Systems
The development of different methods in photocatalysis has made it more important in a core method of modern synthesis. Photocatalysts absorb visible light to excite electrons that can trigger either energy transfer (EnT) or single-electron transfer (SET) processes. Simple examples include [Ru(bpy)?]²?, which can operate as both an oxidant and reductant. Key advantages of photocatalysis are its mildness, chemoselectivity, and compatibility with aqueous and biological conditions. These properties not only used in complex molecule synthesis but also biomedical applications, through which photocatalysis act as a bridge between chemistry and biology.
4.1 Initial Applications – Photodynamic Therapy (PDT).
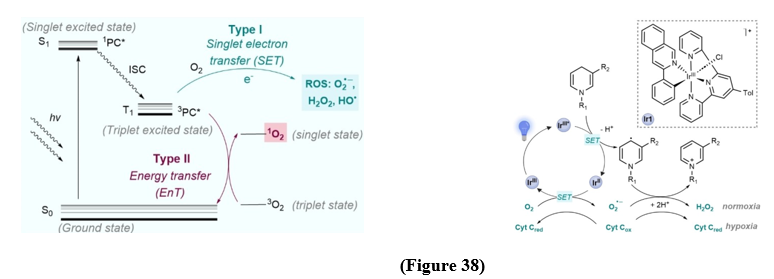
Light-based therapies were used in ancient times that chang into modern PDT where photosensitizers generate reactive oxygen species (ROS) to induce cytotoxicity, mainly in cancer treatment. While porphyrin dyes dominate, metal-based complexes offer higher stability and photophysical properties. Although there are some limitation such as oxygen dependence encourage alternative photocatalytic strategies, including mitochondrial-targeted Ir(III) complexes that oxidize NADH or photoactivated chemotherapy (PACT) (Figure 38). These laid the groundwork for abiotic photocatalysis in biology.[55].
4.2 Net-Oxidative and Net-Reductive Processes.
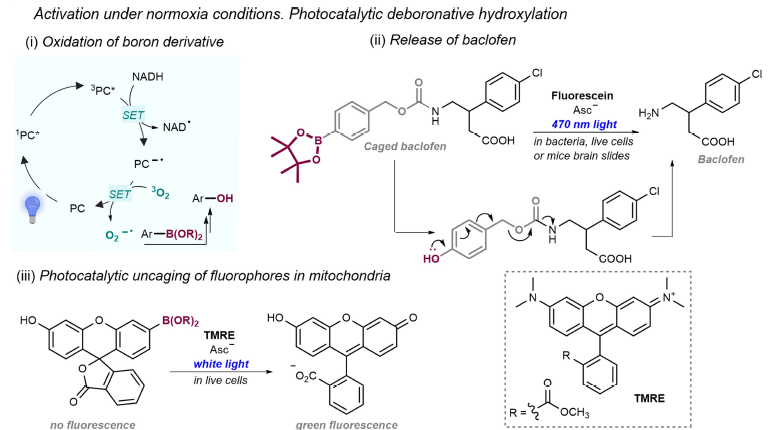
Recent development focus on adapting photoredox chemistry to biological application. Chen’s group developed photocatalytic deboronative hydroxylations for drug uncaging in cells using dyes like fluorescein. Zou and colleagues developed photo-release method for anticancer drugs under hypoxia, while Fox developed far-red tetrazine oxidation for intracellular drug activation. On the reductive side, Ru(II)-catalyzed azide reductions enabled selective uncaging of biomolecules and nucleic-acid templated imaging, later expanded to protein- and RNA-directed photocatalysis with subcellular precision. (Figure 39) [56]
(Figure 39)
4.3. Covalent Biopolymer Modification.
Photocatalysis has been used for bioconjugation, enabling selective labeling and cross-linking of peptides, proteins, and nucleic acids. Kodadek used Ru(bpy)?²? for tyrosine oxidation, while MacMillan developed photoredox peptide cyclizations through decarboxylative radical chemistry. Such methods allow fluorescent tagging, cross-linking, and functionalization of residues including tyrosine, tryptophan, cysteine, and histidine. This can be applicable to live systems, including Fox’s tetrazine-based cross-linking in living mice. (Figure 40) [57]
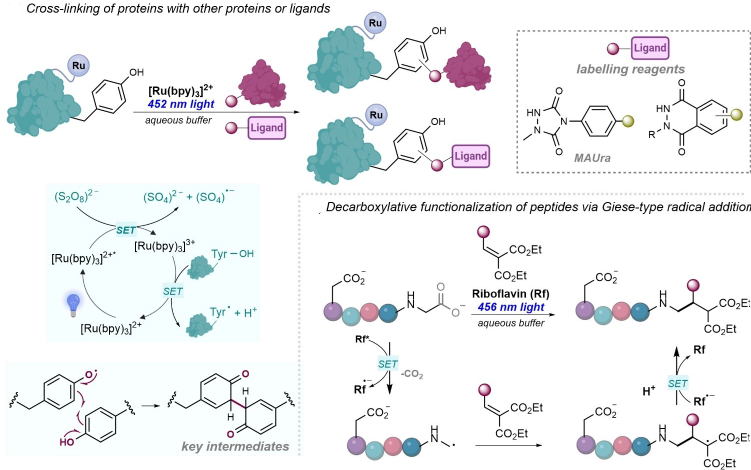
(Figure 40)
4 – CONCLUSION
Photoredox catalysis has become a powerful and greener tool in modern organic synthesis by providing the different methodologies for diverse bond-forming reactions under mild and sustainable conditions. Different developments of photoredox methods discussed in this review highlight how visible-light-mediated concepts have expanded the scope of C–C and C–X bond constructions, including desulfonylation, decarboxylation, defluorinative methodologies, allylic functionalization, and radical-mediated pathways. These all methods not only offer a greener pathway to traditional transition-metal catalysis but also open new avenues for late-stage functionalization of complex bioactive molecules.
Along with synthetic application, photoredox catalysis has greater connections with bioorthogonal chemistry and biological applications, showing its potential in drug discovery, bioconjugation, and photodynamic therapy. In spite of remarkable progress, there are some challenges, such as catalyst design, scalability, energy efficiency, and compatibility with highly functionalized substrates. Future developement in this field focused on naturally available photocatalysts, visible to near-IR light reaction, and connection between chemistry and dual catalysis is expected to further accelerate the adoption of these methods in academics and industry. Although, the continuous development of photoredox catalysis will not only evolve different methods in synthetic chemistry but also expand its applications across pharmaceuticals, materials science, and biological systems.
REFERENCES



Manusiya Shakirali*, Deep Aghara, Recent Advances in Photoredox Catalysis for Organic Synthesis, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 1315-1343 https://doi.org/10.5281/zenodo.17106868
 10.5281/zenodo.17106868
10.5281/zenodo.17106868