
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Chemical Technology, Dr. Babasaheb Ambedkar Marathwada University, Chh. Sambhajinagar, Maharashtra.
Epidermal growth factor receptor (EGFR) is a receptor for epithelial growth factor (EGF) signaling and cell proliferation, which is related to the inhibition of metastasis, tumor cell proliferation, apoptosis, angiogenesis, and tumor invasion. Thus, as a result, it has emerged as a crucial cancer therapeutic target. Unfortunately, after several months of treatment, prior generations of tyrosine kinase inhibitors for EGFR lose their effectiveness due to the rapid emergence of EGFR mutation-mediated resistance (L858R, T790M, or C797S), despite their initial impressive response. There hasn't been a significant development in the study of third-generation inhibitor resistance. In order to target the triple mutant epidermal growth factor receptor with mutation, substantial emphasis has been paid to the development of fourth-generation EGFR-TKIs. This review paper intends to collect references in this field and summarize recent progress in targeting EGFR mutant cancers with specific inhibitors for overcoming the L858R/T790M/C797S triple mutation in cancer patients. It might give immediate information for future structural modification and medication discovery.
Cancer continues to be a cause of death worldwide. Many advances have been made with regard to the lung cancer, especially with regard to targeted therapies, and these treatment strategies have resulted in considerable improvements in survival. Lung cancer is widely categorized as NSCLC (non-small cell lung cancer) and SCLC (small cell lung cancer) (Rathod et al., 2023). The most crucial enzyme is the epidermal growth factor receptor in the tyrosine kinase family (Pezzuto et al., 2023). EGFR is generally a transmembrane receptor located on the surface of the cell. Structural similarities categorize ErbB1 (HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4) are EGFR enzymes that show action when binding with ligand. HER2 and EGFR are well-known targets used in targeted cancer therapy and have been extensively investigated by both academia and industry. The EGFR enzyme triggers numerous downstream signaling pathways (Q. He et al., 2023).
According to a recently reported study, overexpression or abnormal activation of EGFR and ErbB-2 frequently causes malignant cell transformation (Giaccone & He, 2023). These reports make EGFR and ErbB-2 interesting targets for cancer research. Effective targeted therapies are carried out by specially targeting an enzyme or protein that transports mutated or any other genetically modified or altered cells that are different than normal tissue cells (Nakamura et al., 2023). Mutations in enzymes are classified into four different types, termed four generations, as a result of drugs acting on a particular mutated enzyme. The first generation includes erlotinib (2) and gefitinib (1) (Figure 2), which were mostly used extensively with regard to the cancer in the beginning (Liu et al., 2023). However, all patients experience tumor resistance following a median period of response, because of the appearance of the EGFRT790M resistance mutation. This limits the long-term efficacy of these inhibitors. Figure 1 shows the observed mutations in wild type EGFR enzyme. Observed mutations in wild type enzyme, create a need for development of second-generation EGFR inhibitors(Tang et al., 2023).
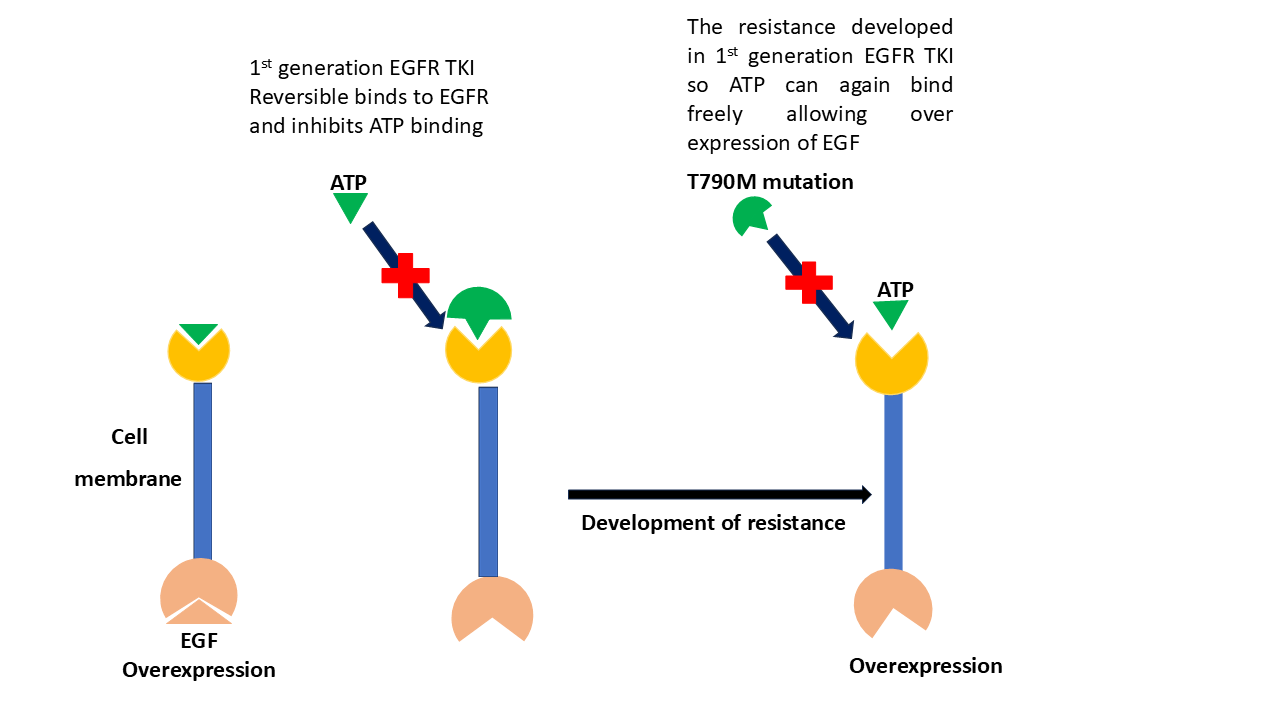
Figure 1. Observed mutations in the EGFR enzyme caused development of resistance in 1st generation tyrosine kinase inhibitors.
Using the quinol Inamine structural motif, the second generation of EGFR-TKIs was also found. Afatinib (3) and Dacomitinib (4) (Figure 2). serve as illustrative examples of these irreversible, non-selective EGFR-TKI inhibitors. A particular functional group known as michael addition receptor is present in the majority of second-generation EGFR-TKIs, for instance, unsaturated amide (Karnik et al., 2021). The sulfhydryl group of Cys797 can be covalently bound by these unsaturated amides to create irreversible inhibition, boosting the affinity for EGFR-TK proteins. EGFR-TKIs of the second generation often lack EGFR selectivity; they inhibit both mutant and wild-type (EGFRWT) EGFR. In tissues or organs like the skin and digestive system, where normal EGFR is not selectively inhibited, this could lead to unfavorable effects like rash and diarrhea. The medication resistance brought on by the T790 M mutation was not considerably reduced by the second-generation EGFR-TKIs. Clinically, it is exclusively applied to individuals who have received reversible EGFR-TKI therapy and have recurrent NSCLC. Due to several drawbacks and toxicity, these molecules were replaced by third generation molecules (Bai et al., 2023)
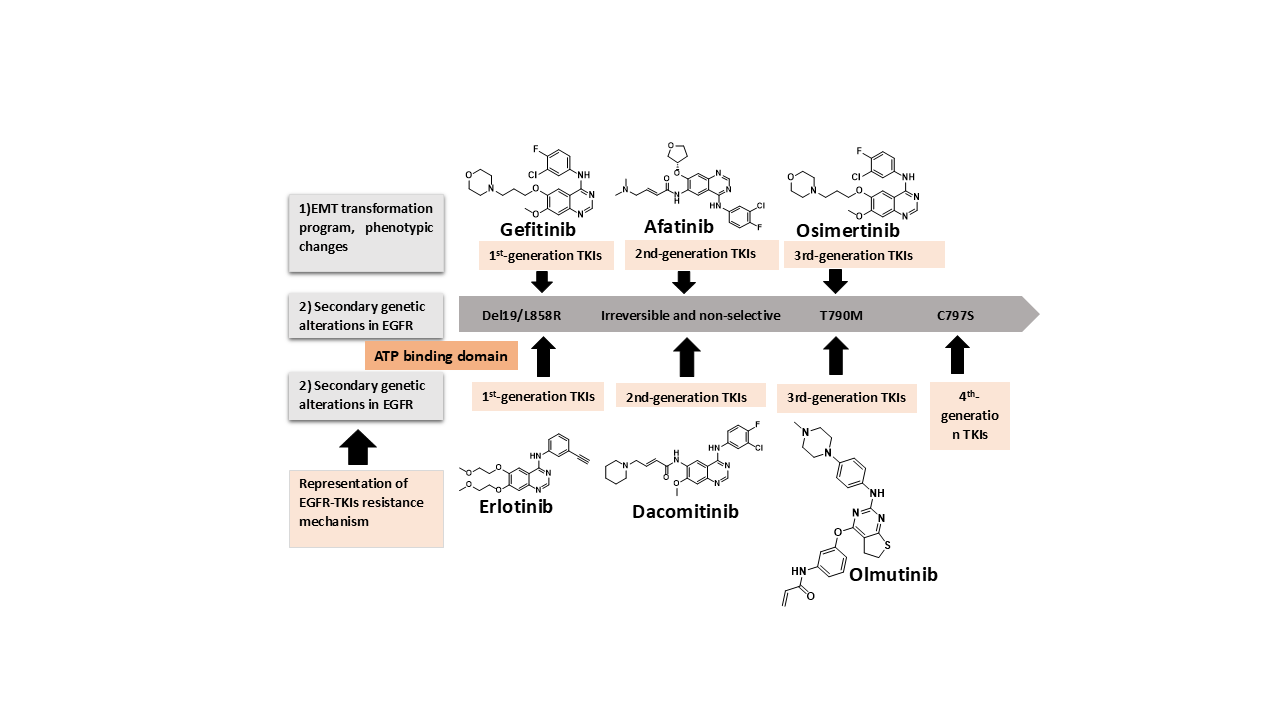
Figure 2. A schematic showing the primary mechanisms of EGFR-TKI resistance and development of three generations of EGFR-TKIs.
Known mechanisms are induction of a transcription program for EMT and phenotypical changes, secondary resistance mutations occurring in the ATP-binding domain (such as T790M and C797S), mutation or amplification of bypass signaling (such as AXL, Hh, ERBb2, etc.), and activating mutations in the downstream pathways (PI3K, AKT, MEK, etc.). EMT, epithelial-to-mesenchymal transition; TKI, tyrosine kinase inhibitor; PI3K, phosphoinositide 3-kinase. The third generation of irreversible EGFR-TKIs is a result of the emergence of new types of drug resistance. Third-generation EGFR-TKIs have pyrimidine as their main structural component; they can bind to the C797 site and target the T790M mutation and EGFR gene-sensitive mutations (exon 19 deletion mutation and L858R mutation) with high specificity (L. Wang et al., 2023). The representative drug is Osimertinib (5, Figure 2) (S. Li et al., 2022). Acquired drug resistance to Osimertinib emerged after administering it to the patients for 10 months, and about 40% of drug-resistant cases were attributed to the C797S mutation. The cysteine 797 residue is absent in the C797S mutant. Because of Michael's addition of the thiol group of C797 to TKIs, the acrylamide warhead of the third-generation EGFR-TKIs can no longer form covalent binding with EGFR mutants, resulting in an undeniable decrease in anticancer efficacy (T. G. Lee et al., 2023). Furthermore, in patients who have the T790M mutation, the C797S mutation is also a potential factor that contributes to resistance to such irreversible EGFR-TKIs as HM61713, WZ4002 and CO-1686. The development of mutations in third-generation molecules was confirmed by monitoring plasma samples pre-treatment and post-treatment. Further, this led to the progress of fourth-generation molecules (Russo et al., 2017). The creation of fourth-generation EGFR-TKIs was required due to the prevalence of the frequently acquired C797S mutation. The triple EGFR mutations L858R/T790M/C797S or Del19/T790M/C797S or their C797S-containing variants have been the subject of intense research efforts in recent years.
In recent years, allosteric drug development has focused on allosteric (targeting tyrosine kinases outside the ATP binding site) inhibitor development, as allosteric regulation is a common occurrence in protein kinases (Leroux & Biondi, 2020). The development of allosteric inhibitors may alleviate some commonly encountered problems like dosage and off-target side effects (De Smet et al., 2014). thus, creating a demand to the finding of novel allosteric inhibitors. Allosteric molecules were searched, which screened wild-type as well as mutant enzymes. Allosteric inhibitor of EGFR or EAI001 was the first compound to possess potency (EGFR L858R/T790M IC50 of 0.024 mM) and selectivity (EGFR wild-type IC50 > 50 mM) for mutant EGFR. Nevertheless, it was only moderately effective against isolated T790M and L858R mutations. Due to the inclusion of an electronegative fluorine group, the EAI045 inhibitor was discovered to have significant efficacy and selectivity for the L858R/T790M mutation following medicinal chemistry-based optimization of EAI001. Profiling a panel of 250 protein kinases revealed the EGFR mutant to be very selective. As a result, it was determined that EAI045 inhibits mutant EGFR in an allosteric, non-ATP-competitive manner. Targeted cancer therapies are anticipated to be less harmful to healthy cells and more effective than more traditional cancer therapies (Ferrara et al., 2023). The most successful targeted therapies are chemical agents that specifically or preferentially target a protein or enzyme that bears a mutation or other genetic alteration that is particular to cancer cells. There are targeted therapies for colorectal cancer, head and neck cancer, breast cancer, multiple myeloma, lymphoma, prostate cancer, melanoma, and other cancers (Patel et al., 2017).
EGFR AND ITS SIGNALLING PATHWAYS (DOWNSTREAM): The epidermal growth factor (EGF) receptor is a transmembrane receptor (also known as the HER1 or ErbB1 receptor) has tyrosine kinase activity. It has an internal protein tyrosine kinase domain that binds ligands and a C-terminal, an extracellular protein tyrosine kinase domain, a hydrophobic transmembrane domain, a juxta membrane domain. It usually exists as either monomers (which are not active) or dimers (which are active), and both are ready to bind to extracellular ligands and start a number of cascades (Kardile et al., 2023). However, some EGFR mutations have the ability to activate the protein constitutively even when its ligand is not present. These mutations, which are gain-of-function mutations, can be categorized into rare mutations and traditional activating mutations (exon 19 deletion mutation, exon 21 L858R mutation, etc.). Together, the L858R point mutation in exon 21 and the exon 19 deletion make up around 85% of all EGFR mutations in NSCLC. (Brea & Rotow, 2023). These two variants can be categorized as classic activating EGFR mutations since they are both considerably impacted by the sensitivity of the first-generation EGFR-TKI. Comparatively, rare EGFR mutations have a low mutation rate, making up only 15% of all EGFR mutations. These alterations include point mutations, deletions, and insertions in exons 18–25 of the EGFR gene. (Uribe et al., 2021). Since EGFR mutation sites are frequently found throughout the ATP-binding site of its kinase, any subsequent changes to the amino acid residues in this region will change the chemical features of the bound place (hydrophobic contact, creation of hydrogen bonds, etc.). This will disrupt the kinase's affinity for ATP and the stability of its inactive conformation, which will then result in the aberrant activation of its kinase activity (Alharbi et al., 2022).
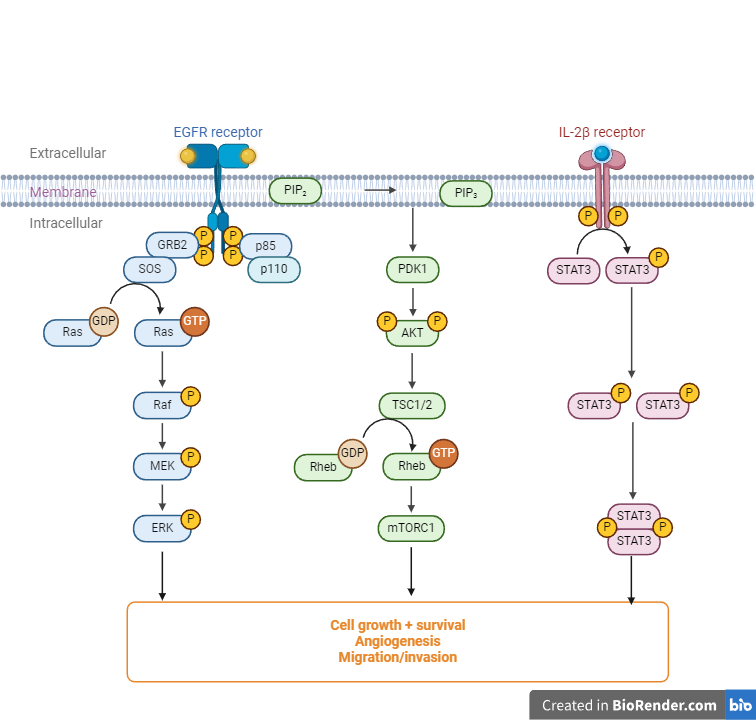
Figure 3. EGFR signalling green and purple spheres indicate the EGFR-activating ligands.
As a result, EGFR gain-of-function mutations frequently act as drivers of carcinogenesis, activating downstream EGFR signaling independent of ligands, or occasionally as drivers of drug resistance, rendering TKIs ineffective (Damghani et al., 2023). Whether EGFR is activated naturally or abnormally, it will nonetheless lead to the activation of numerous downstream signaling cascades (Hayashi et al., 2022). The first downstream signalling pathway is RAS-RAFMEK-ERK pathway. After ligand binding, EGFR dimerizes, activating RAS, which then causes RAF kinase to phosphorylate MEK. Phosphorylation of MEK sequentially triggers ERK activation, which leads to the activation of cell cycle-related transcription-related variables like C-FOS, MYC, CREB, etc. These transcription factors further initiate the transcription of target genes, such as cyclin D, and eventually promote cell division, proliferation, invasion, and metastasis. The PI3K/AKT signal route is the second signaling pathway. AKT, a key oncogenic hub molecule, is activated when PIP-3, a phosphatidylinositol 3, 4, and 5-triphosphate molecule, is stimulated by the phosphorylated EGFR tyrosine kinase. The expression of associated proteins necessary for cell cycle progression from the G1 phase to the S phase and elevation of the anti-apoptotic cascade signals, among other things, are then upregulated once mTOR, the important downstream regulatory molecule of AKT, is activated. The JAK/STAT signal transduction system, which primarily controls immunity and mediates the proliferation and invasion of cancer cells, is last but certainly not least. These signal transduction pathways have their respective functions and play important roles in enhancing mitosis, cell growth, and transcriptional activation, apoptosis, invasion, and metastasis (Bhattacharya, 2023). EGFR molecular domains contain domains of human EGFR and exons encoding them. It is located on chromosome 7, which has 28 exons. EGFR-dependent drug resistance mutation sites include the T790 M’ gatekeeper’ mutation, the L858R mutation, tertiary mutations such as C797S, and EGFR molecular structure (Tripathi & Biswal, 2021).
Development Of the Fourth?Generation TKI:
Considering that the C797S mutation is the common cis configuration (85%), researchers and drug companies are working hard to find ways to get around the C797S-based resistance to the third-generation TKIs. According to Table 1, the development of fourth-generation TKIs and combination therapy are two potential solutions to this issue. The latter gained attention in particular because all currently available fourth-generation TKIs, including EAI045, brigatinib, CH7233163, etc., showed promising curative outcomes in NSCLC patients carrying the C797S mutation. (Y. Li et al., 2023). For example, brigatinib (Alunbrig), a novel dual kinase inhibitor of ALK and EGFR, was approved by the FDA in April 2017 (Zhao et al., 2022) and utilized as the first line of treatment for individuals with locally progressed or metastatic ALK-positive NSCLC. It effectively inhibits the growth of ex19del-T790M-C797S triple mutant cells both in vitro and in vivo, and it performs even better when combined with the anti-EGFR antibody cetuximab. Wang et al. reported that a Geftinib and later osimertinib were used to treat the patient's EGFR ex19del-mutated lung adenocarcinoma at first, but the illness progressed after acquiring a triple mutation of EGFR ex19del/T790M/cis-C797S and used cetuximab along with brigatinib to obtain considerable effectiveness. The objective response rate (ORR) for 15 patients with osimertinib resistance who also had the mutations C797S and T790M was 60% following treatment with the drug. The disease control rate (DCR) when brigatinib and cetuximab were combined was 100%. To successfully treat lung adenocarcinoma with the triple mutation of the EGFR, L858R-T790M-cis C797S, brigatinib can also be used in combination with the VEGF inhibitor bevacizumab in addition to anti-EGFR antibodies. In addition to brigatinib, Yong Jia’s team found that the combination of the novel EGFR allosteric inhibitor EAI045 with cetuximab has a significant effect on the L858R-T790M-C797S triple mutant but not on the ex19del mutant NSCLC cells. Fortunately, the EGFR triple mutation ex19del-T790M-C797S was successfully inhibited by a different fourth-generation TKI, CH7233163. Despite the fact that an excellent fourth-generation TKI has yet to be developed, the future is as more and more applicants with improved therapeutic benefits and pharmacokinetic and pharmacodynamic features are appearing. (Xu et al., 2023)
Table 1. The fourth-generation EGFR-TKIs in clinical trials
|
Inhibitor |
Status |
NCT identifier |
Sponsor |
|
BLU-945 |
Phase I |
NCT04862780 |
Blueprint Medicines |
|
BLU-701 |
Phase I |
NCT05153408 |
Blueprint Medicines |
|
TBQ3804 |
Phase I |
NCT04128085 |
Chia Tai Pharmaceutical Group Co., Ltd |
|
BBT-176 |
Phase I |
NCT04820023 |
Bridge Biotherapeutics |
|
BPI-361175 |
Phase I |
NCT05329298 |
Betta Pharmaceuticals Co., Ltd. |
Figure 3. Approved EGFR-TKIs
SMALL MOLECULAR EGFR-TKIS: The EGFR C797S mutation in NSCLC therapy resulted in drug resistance to osimertinib, which led to the identification of various novel 4th generation EGFR inhibitors. By using X-ray crystal diffraction, the interaction mode between the inhibitor and the EGFR C797S mutant could be demonstrated, which should help with future structure modification. In order to give the community a reference for clinical NSCLC therapy, the most recent research developments on various categories and chemical scaffolds of small molecule inhibitors were reviewed in this article and to accelerate the drug discovery in this field (Johnson et al., 2023).
EAI001: In In 2016, Jia et al. reported allosteric inhibitors that overcome drug resistance due to EGFRT790M and EGFRC797S mutations. They analyzed a library of 2.5 million compounds and They discovered EAI001 (Figure 4) as a promising non-ATP-competitive inhibitor and selective inhibitor against EGFRL858R/T790M. X-ray crystallographic data analysis proved that EAI001 forms at the allosteric site, close to the ATP binding site of the T790M-mutant EGFR. EAI001 was found to bind specifically to the Aminothoazole and mutant portions of the EGFR, and to create hydrogen bonds with the NH-group of the carboxamide and the Asp855 units. With an IC50 of 24 nM (at 1 M ATP), EAI001 demonstrated superior potency, selectivity, and specific binding capacity for mutant EGFRL858R/T790M compared to wild-type EGFR (IC50 > 50 M). A more effective EAI045 against EGFRL858R/T790M was produced by further optimizing the EAI001 structure (IC50 = 3 nM). (J. He et al., 2021).
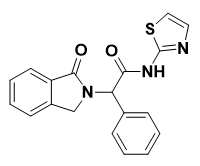
Figure 4. Structure of EAI001
EAI045: A library screening of about 2.5 million compounds with specific EGFR mutant selection led to the discovery of EAI001, a new allosteric inhibitor of EGFR with a thiazole amide skeleton. The IC50 value of EAI001 against the T790M and L858R mutant EGFR kinases reached 24 nM, while the IC50 value for the EGFRWT was more than 50 mM. EAI045 emerged from the optimizing EAI001 using medicinal chemistry (Figure 5). At a concentration of 1 mM ATP, the IC50 of EAI045 to T790M/L858R mutant kinase was 3 nM, hence being more than 1,000-fold selective when compared with EGFRWT, in which the inhibition is not affected by the concentration of ATP. According to the the EAI001 crystal structure binding to T790M-mutant EGFR, EAI045 may also bind to allosteric sites in the EGFR kinase domain and require EGFR to stay in the C-helix-out conformation (Gero et al., 2022). The mechanism driving the selectivity of EAI045 toward mutant EGFR was identified by molecular dynamics simulations. Leu858's side chain is persistently buried in the allosteric pocket that is hydrophobic in EGFRWT, which prevents EAI045 from binding to EGFR. In the L858R mutant of EGFR, Arg858 induces the allosteric pocket to open, thus enabling the binding of EAI045 to EGFR. Other mutations with a similar structure that might expose the allosteric pocket, including L861Q, might also have a strong affinity for EAI045. However, EAI045 exhibited poor antitumor activity and incomplete inhibition of EGFR autophosphorylation in cancer cells. In EGFRWT asymmetric dimers, the "activators " C-lobe subunit changed the in shape of the C-helix on the N-lobe of "receiver" subunit. Hence, only the "receiver" subunit is activated in EGFRWT. In mutant EGFR dimers, both subunits could activate the downstream signalling pathway while EAI045 only binds to one subunit, resulting in incomplete inhibition of mutant EGFR. They could increase the effectiveness of EAI045 when combined with other drugs that could prevent EGFR dimerization. More precisely, cetuximab effectively enhanced the activity of EAI045 toward L858R/T790M/C797S triple EGFR mutant cells(S. Wang et al., 2017). Even though the T790M mutation of EGFR without L858R showed a decreasing trend as the ATP concentration went up, EAI045 still showed a promising ability to block the T790M mutation. However, the mentioned molecular dynamics simulations could not explain this robust EAI045 inhibition of the T790M/C797S EGFR mutation. The crystal structure of EAI045T790M/C797S/V948R mutant EGFR indicated that EAI045 could bind to a deep pocket between the ATP pocket and the C-helix, thus requiring adequate space created by the C-helix-out rotation. Amino acid deletion pulls the C-helix toward the ATP-binding pocket and constrains the allosteric site to a small volume, leading to the low affinity of EAI045 for the Ex19del EGFR mutation. However, EAI045 is inactive as a solitary agent and is only active when combined with EGFR-targeted mAbs, such as cetuximab. Cetuximab’s on-target EGFRWT toxicities may limit its clinical translational potential (K. Wang et al., 2023).
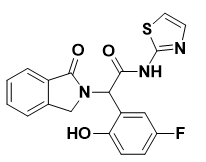
Figure 5. Structure of EAI045
BDTX-1535: Lucas et al. have studied BDTX-1535. BDTX-1535 (Figure 6), a fourth-generation EGFR inhibitor, targets mutations causing intrinsic and acquired resistance in NSCLC. Epidermal growth factor receptor (EGFR) mutations in exon 19 del and exon 21 (L858R) are the two most common mutations in NSCLC. Osimertinib, a third-generation tyrosine kinase inhibitor for EGFR (TKI), has emerged as the preferred first-line therapy for NSCLC patients carrying these mutations. Third generation TKI resistance can be caused by secondary changes to EGFR, such as a change from cysteine to serine at position 797 (C797S). Real-world evidence shows that, in addition to C797S, EGFR changes like kinase domain mutations (like S768I), extracellular domain changes (like EGFRvIII, A289X), and EGFR amplification also happen when osimertinib is used to treat cancer. In addition to the classical mutations, NSCLC tumors have a wide range of primary kinase domain mutations, such as G719X in exon 18, S768I in exon 20, and L861Q in exon 21, that make them resistant to approved TKIs (Iyer et al., n.d.). NCCN guidelines say that afatinib or osimertinib should be used. However, there is still a need for an EGFR TKI that targets these mutations and is highly effective, well tolerated, and can get into the central nervous system. Our MAP platform let us choose MasterKey inhibitor BDTX-1535, which is a CNS-penetrating EGFR TKI of the fourth generation that targets multiple intrinsic and acquired EGFR resistance alterations in a specific and potent way (W. Wang et al., 2023). The effects of BDTX-1535 on different species were studied before it was used in people, and PK and Kpuu values were found in the brain and plasma. Numerous mouse PDX and transplant models were used to evaluate antitumor activity. The alterations in Exon 18–21 that are associated with innate or learnt resistance to 3rd generation EGFR inhibitors are the target of BDTX–1535, an effective and selective CNS–penetrant, irreversible EGFR TKI. It can get into the central nervous system without harming wild-type cells. These include a wide range of EGFR mutations inside the kinase domain (such as C797S, L718Q, G724S, and S768I), extracellular domain changes (such as EGFRvIII and A289X), and EGFR amplification. BDTX-1535 consistently causes tumors with these mutations to regress at well-tolerated doses without causing appreciable EGFR wild-type-associated toxicities based on research using mice xenograft and allograft. BDTX-1535 is active in an intracranial PDX model and orally accessible with a CNS Kpuu of 0.55 in rats and 0.48 in dogs, respectively. The MAP platform allowed for the discovery of BDTX-1535, a fourth-generation CNS-penetrant EGFR TKI that permits MasterKey targeting of EGFR mutations linked to intrinsic or acquired resistance to third-generation EGFR TKIs while sparing wild-type EGFR. Due to its extensive coverage of resistance mutations, BDTX-1535 can address a large unmet medical need in EGFR mutant lung cancer and CNS penetration capabilities. Phase I clinical research on BDTX-1535 is now underway (NCT05256290). ("BDTX-1535 Goes After Osimertinib Resistance," 2021).
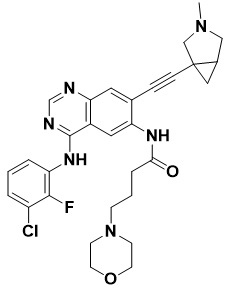
Figure 6. Structure of BDTX-1535
JIN-A02: M.R. Yun et al. have studied JIN-A02. EGFR C797S mutations in NSCLC are the focus of JIN-A02, a fourth-generation, highly efficient tyrosine kinase inhibitor with intracranial action. EGFR tyrosine kinase inhibitors (TKI) have improved treatment results for EGFR-driven NSCLC, although resistance always develops and the illness frequently worsens as a result of brain metastases. The most significant resistance mutation to develop following therapy with a third-generation TKI is C797S. JIN-A02 is an EGFR-TKI with good brain penetration that is accessible orally and targets the C797S mutation. (Izumi et al., 2022). By measuring phosphorylation-EGFR expression using the AlphaLISA assay and using patients' derived cells (PDC) and patients' derived organoids (PDO) on EGFR mutants, the cellular activities of JIN-A02 were assessed. By using whole-exome sequencing, various allelic associations of various sorts were also evaluated in all models. Patients-derived xenografts (PDX) were used to assess the antitumor activities of JIN-A02. A tumor xenograft produced from the NCI-H1975 cell line was employed for in vivo intracranial activity. The AlphaLISA assay revealed that JIN-A02 was highly effective in EGFRex19del/T790M/C797S (IC50 = 4.7 nM, Ba/F3) and EGFRL858R/T790M/C797S (IC50 = 12.8 nM, NCI-H1975 LTC). JIN-A02 significantly reduced cellular activity in the trans model (IC50=1489.7 nM, PDO) and in the cis model (IC50=92.1 nM, PDC) of EGFRex19del/T790M/C797S. With an IC50 of above 4,000 nM for both cells, it seems to be superior to osimertinib. JIN-A02 also demonstrated similar potency to osimertinib in EGFRex19del/T790M (IC50 = 84.4 nM for PC-9GR and EGFRL858R/T790M; IC50 = 73.3 nM for NCI-H1975) and EGFRL858R/T790M (IC50 = 73.3 nM for NCI-H1975). JIN-A02 slowed tumor growth in the PDX mouse model (YHIM-1094, EGFRex19del/T790M/C797S), when administered at a dose of 30 mg/kg. JIN-A02's anticancer activity was demonstrated in the brain metastases model with intracranial implanted NCI-H1975 tumors. JIN-A02 exhibits intracranial action and is a very effective EGFR-TKI for a variety of EGFR targeted mutations, including cis and trans EGFRex19del/T790M/C797S. JIN-A02 is anticipated to offer a possible treatment possibility for NSCLC based on their strong actions for EGFR mutations. (Mezquita et al., n.d.).
CH7233163: A huge chemical library was used to find CH7233163 (IC50 = 0.28 nM), a new compound that can defeat the resistance brought about by the Ex19del/T790M/C797S triple mutation (Figure 7). CH7233163 blocked a wider range of EGFR mutations than EGFRWT, like L858R/T790M/C797S, L858R/T790M, Ex19-del/T790M, Ex19del, and L858R. Additional experiments confirmed CH7233163's strong anticancer efficacy in tumor cells with the triple mutations Ex19del/T790M/C797S or L858R/T790M/C797S. It was discovered from the structure of crystals of the triple mutant EGFR CH7233163-L858R/T790M/C797S that CH7233163 interacts with the ATP-binding pocket via hydrogen bonds and CH/ interactions but not with the Ser797 residue. Compared with osimertinib, CH7233163 not only binds to the P-loop and the hinge region but also directly interacts with Met790 residues. Compared with EAI001, CH7233163 binds to C-helix of the conformation of EGFR, contributing to its high affinity toward the Ex19del/T790M/C797S mutation (Du et al., 2021).
Figure 7. Structure of CH7233163
JBJ-04-125-02: A novel molecule, JBJ-04-125-02, was created by using an iterative strategy for the synthesis of structural analogs of EAI001 (Figure 8). The crystal structure demonstrated that JBJ-04-125-02 interacts to the allosteric pockets in the EGFR C-helix-out conformation just like EAI045 does. It's interesting to note that JBJ-04-125-02 binding caused the A-loop to adopt a unique conformation that appears to be supported by a hydrogen connection between the piperazine group of the molecule and Glu865 in the structure. Without the coadministration of cetuximab, JBJ-04-125-02 had a cytotoxic impact on the L858R, L858R/T790M, or L858R/T790M/C797S mutations. JBJ-04-125-02 also lacks binding affinity toward EGFRWT or the Ex19del mutant. Further research indicated that osimertinib might enhance the binding affinity of JBJ-04-125-02 to EGFR, leading to more potent antitumor activity. These results indicated that some lung cancer patients may be resistant to EGFR-TKIs of the third generation may benefit from combining a covalent mutant-selective allosteric EGFR-TKI with other forms of therapy. (Mai et al., 2023).
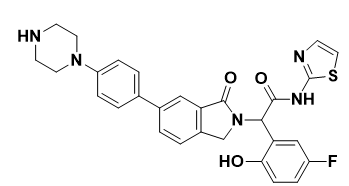
Figure 8. Structure of JBJ-04-125-02
BLU-945: Another fourth-generation EGFR-TKI medication is BLU-945 (Figure 9) developed by Blueprint Medicines that targets the T790M/C797S mutation reported in ESCO and other T90M resistance mutations secondary to osimertinib resistance. BLU-945 has been shown to have enzyme activity against EGFRT790M/C797S with >900-fold selectivity over wild-type EGFR (IC50 = 0.5 nM). In the NCI-H1975 CDX (L858R/T790M) model, BLU-945 was shown to be efficient and comparable to osimertinib. In vitro data suggested that BLU-945 achieved robust inhibition of L858R/T790M/C797S and Ex19del/T790M/C797S triple mutations rather than EGFRWT. Cell-derived xenograft and patient-derived xenograft models consistently showed that single treatment with BLU-945 or coadministration with osimertinib or gefitinib significantly blocked the progression of tumors with the Ex19del/T790M/C797S mutation. Brigatinib was identified as a next-generation inhibitor targeting anaplastic lymphoma kinase (ALK). A series of screening studies suggested that brigatinib potently inhibits the proliferation of cells harboring the triple EGFR mutation. Moreover, several small-molecule compounds, including the 4-aminopyrazolopyrimidines, tri-substituted imidazoles, and 2-aryl-4-amino quinazolines, have also been reported to be able to surmount C797S mutation-mediated drug resistance. Additional clinical trials for these fourth-generation EGFR-TKIs may offer a promising therapeutic strategy to overcome mutant EGFR-based TKI resistance in various cancers (Eno et al., 2022).
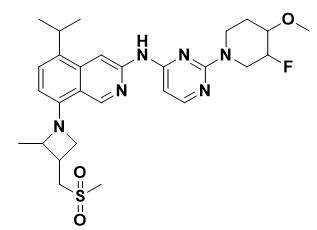
Figure 9. Structure of BLU-945
TREA0236: TREA-0236, another allosteric inhibitor, was created by Lee et al. (Figure 10), by replacing aminothazole with quinazoline-4-one on EAI045. But, TREA-0236 (IC50=5.3 μM) was not as effective as EAI045 (S. Lee et al., 2018).
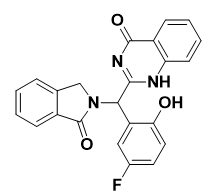
Figure 10. Structure of TREA0236
BI-4020: At Boehringer Ingelheim, Engelhardt et al. screened a molecule library and identified the aminobenzimidazole compound (Figure 11) as a promising hit. The drug demonstrated good selectivity in 238 kinase tests and resistance to EGFRL858R/T790M/C797S (IC50 = 2100 nM) and EGFR19Del/T790M/C797S (IC50 = 250 nM). At the ATP binding pocket, the inhibitor was bonded to the mutant protein. They altered the core, rigidified it through macrocyclization, and discovered another superior molecule, BaF3, to increase the efficacy. When compared to an open-chain molecule, the cyclic structure's biological activity increased 17 times. BaF3 cell lines were used to assess BI-4020's antiproliferative properties. The triple mutant EGFR19Del/T790M/C797S (IC50 = 0.2 nM) was effectively active by BI-4020 and was selective over EGFRwt (IC50 = 190 nM). The effectiveness of BI-4020 in triple mutant EGFR19Del/T790M/C797S NSCLC xenografts was shown. (Goel et al., 2023).
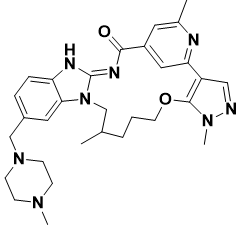
Figure 11. Structure of BI-4020
BRIGATINIB: Brigatinib (Figure 12) is an FDA-approved kinase inhibitor to treat patients with metastatic ALK-positive NSCLC. Brigatinib has recently been demonstrated to be effective in reducing and significantly suppressing the kinase as well as inhibiting the growth of EGFRC797C-mutated cell lines. (Fang et al., 2022). Because there was not enough brittinib in the plasma, it was ineffective for demonstrating the EGFRC797S mutation in vivo. (Abourehab et al., 2021).
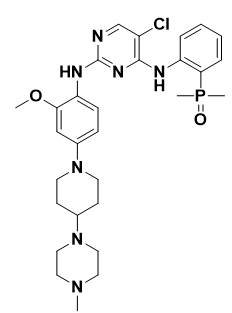
Figure 12. Structure of Bringatinib
JND3229: A pyrimidopyrimidinone derivate named JND3229 (Figure 13) was discovered by Lu et al., as extremely selective and potent EGFRC797S inhibitor (IC50 = 5.8 nM). With IC50 values of 0.51 and 0.32 mM, respectively, JND3229 inhibited the growth of Ba/F3 cells that carried EGFR mutations, specifically EGFRL858R/T790M/C797S and EGFR19Del/T790M/C797S. JND3229 showed good in vivo mono-drug efficacy in xenograft mouse models for EGFR19Del/T790M/C797S. (Das, 2022).
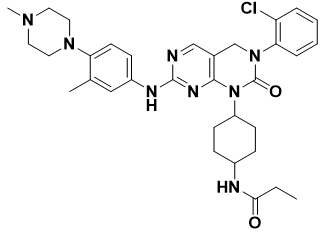
Figure 13. Structure of JND3229
Other compounds, pharmaceutical companies have discovered two new fourth-generation EGFR-TKIs: TQB3804 (Figure 14) and BBT-176. (Lim et al., 2023). Pre-clinical research demonstrated that TQB3804 and BBT-176 displayed a remarkable inhibitory impact on Ex19-del/T790M/C797S and the L858R/T790M/C797S triple mutations.
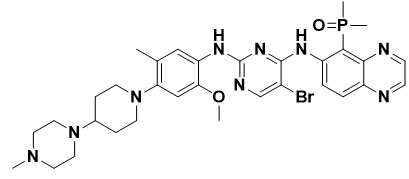
Figure 14. Structure of TQB3804
Table 2: Limitations of first- to fourth-generation EGFR-TKIs with approved drugs.
|
Generation |
Name |
Limitations/failure |
|
First-generation EGFR-TKIs |
Erlotinib Gefitinib Icotinib |
Dysregulated EGFR signaling (abnormalities in IGF1R, HER2, MET, FGR bypass signaling, and HER2 amplification) and changes in downstream EGFR signaling (BRAF mutation, K-RAS mutation, loss of PTEN, and PIK3CA mutation) are linked to T790M mutation-related resistance emanation. The use of first-generation EGFR-TKIs was constrained by the fact that targeting wild-type EGFR causes severe adverse effects and dose-dependent toxicity in patients. |
|
Second generation EGFR-TKIs |
Dacomitinib Neratinib |
Patients who had previously received first-generation EGFR-TKIs did not exhibit an improvement in their overall survival during the clinical trial of phase II. Effective and significant results were not observed during the phase II clinical trial in EGFR-mutated NSCLC patients. |
|
Third generation EGFR-TKIs |
Afatinib Nazartinib Naquotinib Olmutinib Rociletinib Osimertinib |
During clinical trials, no change in the overall survival of patients was observed. Resistance emanating from EGFR-dependent mechanisms such as the emergence of EGFR mutations (L692V, C797G, C797S, G796, L718, L792, L798, and E709K mutations) and EGFR-dependent processes include dysregulated EGFR signaling (MET amplification, FGR bypass signaling, HER2 amplification, HGF overexpression, and anomalies in the IGF1R), as well as mutant EGFR downstream. |
|
Fourth generation EGFR-TKIs |
EAI001 EAI045 DDC4002 DDC-01-163 |
Still not approved at the clinical level and not significantly effective as a cancer preventative alone in preclinical studies. |
CONCLUSION: The tertiary mutation of EGFR significantly restricts the clinical use of third-generation EGFR-TKIs despite their remarkable efficacy. Impairing the covalent attachment of the third-generation EGFR-TKIs with EGFR-TK, one of the vital mechanisms is the C797S mutation for acquired drug resistance against third-generation EGFR-TKIs. EGFR-TKIs alone or in combination cannot suppress disease progression when C797S and T790M alleles occur in cis-form. A number of fourth-generation EGFR-TKIs have been reported to combat drug resistance brought on by the C797S mutation, including ATP-competing and allosteric inhibitors using a range of scaffolds. According to the published research, the allosteric inhibitor EAI045 and cetuximab work in concert to treat non-small cell lung tumors with acquired resistance mutations. According to various patents and scientific studies, brigatinib and its analogues have potential action against triple mutant EGFR with low nanomolar IC50 values. The in vivo antitumor activity also validated their potential for clinical use. Besides, brigatinib appeared to be potent against both EGFRL858R/T790M/C797S and EGFRdel19/T790M/C797S. As a result, this group of inhibitors is well suited to act as the starting point for the creation of fourth-generation EGFR-TKIs. The study is supported by innovative drug discovery and design approaches like library generation, virtual screening, molecular docking, relative binding free energy calculations, ADMET studies, and molecular dynamic simulations in identifying potential lead compounds for the recent fourth-generation C797S mutant EGFR-specific enzyme.
DECLARATION OF COMPETING INTEREST: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
ACKNOWLEDGEMENT: My sincere gratitude goes out to the late Dr. Aniket Sarkate sir, whose advice, expertise, and support profoundly influenced our research. The authors are thankful to The Head, Department of Chemical Technology, Dr. Babasaheb Ambedkar Marathwada University, Chh. Sambhajinagar 431004 (MS), India, for providing the laboratory facility. The author Mahadevi V. Kendre is thankful to the Mahatma Jyotiba Phule Research & Training Institute (MAHAJYOTI), Nagpur 440020 (MS), India, for providing Research Fellowship under Mahatma Jyotiba Phule Research Fellowship-2022 (MJPRF-2022) under Grant MAHAJYOTI/2022/Ph.D.Fellow/1002(462)
REFERENCES


Mahadevi V. Kendre*, Dr. Sachin S. Bhusari, Dr. Pravin S. Wakte, Recent Advances of The Novel Fourth Generation EGFR Inhibitors in Overcoming Triple Mutation for Cancer Therapy, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 1859-1881 https://doi.org/10.5281/zenodo.17140699
 10.5281/zenodo.17140699
10.5281/zenodo.17140699