1,2 Rungta Institute of Pharmaceutical Sciences
3 Rungta Institute of Pharmaceutical Sciences & Research
Cigarette smoke is often described as a complex mixture of toxic compounds; however, its biological impact cannot be understood solely through descriptive toxicology. The present review reconstructs smoke-induced pathogenesis as a deterministic, multi-scale cascade that begins with quantum-level electron transfer and culminates in systemic organ dysfunction. At the molecular interface, redox-active constituents initiate ultrafast radical reactions that disrupt epithelial antioxidant buffering and destabilize mitochondrial electron flow. This disturbance propagates into a coupled collapse of redox homeostasis and ATP bioenergetics, establishing a self-amplifying feedback loop between oxidative stress and metabolic failure. The resulting intracellular environment favors inflammasome activation, persistent cytokine signaling, and progressive genomic instability, including oxidative DNA lesions and epigenetic remodeling. Rather than treating oxidative stress as an isolated phenomenon, this review frames cigarette smoke exposure as a systems-level perturbation of the redox–bioenergetic axis. We integrate mechanistic toxicology, mitochondrial physiology, immunometabolism, and transcriptional regulation into a unified conceptual model that connects femtosecond radical chemistry to chronic diseases such as COPD, atherosclerosis, and lung cancer. Particular emphasis is placed on the Nrf2–NF-?B regulatory balance, the ATP–redox nexus, and the inflammasome–P2X7–mitochondrial ROS convergence as central control nodes of disease progression. The therapeutic implications of this framework challenge the traditional antioxidant paradigm. Direct radical scavenging has failed clinically because it ignores the signaling function of reactive species and the catalytic nature of endogenous redox amplification. Instead, precision strategies must focus on restoring network stability—through modulation of mitochondrial function, redox-sensitive transcriptional programs, purinergic signaling, and systems-informed interventions. By unifying physicochemical initiation, cellular network collapse, and translational modeling within a single continuum, this review proposes a mechanistic foundation for predictive toxicology and network pharmacology. Cigarette smoke–induced disease is not merely cumulative damage; it is a temporally structured systems failure that can be quantitatively modeled, stratified, and potentially intercepted before irreversible organ decline occurs.
Cigarette smoke presents itself as an unusual complex biological challenge. Rather than behaving as a simple toxic exposure, accumulated evidence suggests that it acts as a system-wide perturbation capable of destabilizing cellular order across multiple levels of organization. A recurring theme emerging from decades of research is oxidative stress, which appears repeatedly as the common mechanistic denominator linking smoke exposure to tissue injury. In toxicological terms, oxidative stress reflects a state in which oxidant production overwhelms antioxidant defenses, leading to sustained molecular damage (Boots et al., 2009). It remains intellectually compelling is not the existence of this imbalance which is well established but such a diverse exposure consistently converges on this single pathological axis. To explore this convergence, it is instructive to step beyond descriptive biology and consider the physical constraints governing living systems. Schrödinger’s formulation of life as an open system maintaining order by resisting entropy provides a useful conceptual anchor (Schrödinger, 1944). Within this framework, oxidative stress can be interpreted as a measurable failure of this resistance, a shift toward disorder that manifests biochemically as redox imbalance.
The exposome framework strengthens the line of reasoning by situating oxidative stress within a lifetime continuum of exposure (Wild, 2005). Importantly, the exposome does not distinguish sharply between external insults and internal responses; instead, it treats inflammation, metabolic stress, lipid peroxidation, and redox dysregulation as internalized records of environmental interaction (Wild, 2012). From this viewpoint, cigarette smoke initiates a process that the organism itself propagates, raising the possibility that much of the long-term damage arises from endogenous amplification rather than direct chemical toxicity alone. This idea resonates strongly with the historical evolution of free radical biology. Harman’s Free Radical Theory of Aging reframed reactive oxygen species as causal agents of aging and disease, rather than incidental by-products of metabolism (Harman, 1956). His later identification of mitochondria as a primary source of these species further complicated the narrative by demonstrating that oxidative stress is inseparable from normal cellular respiration. A logical implication follows: cigarette smoke may not introduce an entirely foreign pathology, but instead intensifies an intrinsic vulnerability already embedded within cellular energy metabolism.
Physicochemical analyses of cigarette smoke support interpretation with thousands of reactive constituents, smoke functions less as a single insult and more as a dynamic oxidant field (Boots et al., 2009). Empirical evidence consistently demonstrates a biphasic pattern of injury initial damage from exogenous oxidants followed by a secondary wave driven by activated inflammatory cells releasing reactive oxygen and nitrogen species. Behavioral observations add another layer of complexity. Although smoking is commonly perceived as stress-relieving, physiological measurements indicate that smokers exhibit higher baseline stress levels than non-smokers (Parrott, 1999). The transient relief reported by smokers appears to reflect the suppression of withdrawal-induced stress rather than genuine relaxation. This creates a self-reinforcing loop in which psychological dependence sustains biological stress, suggesting that neurobehavioral dynamics may be inseparable from oxidative pathology.
At the population level, the consequences of this multi-layered interaction are unambiguous. Environmental pollution remains the leading environmental cause of premature mortality, accounting for millions of deaths annually (Landrigan et al., 2018). Within this burden, smoking-related chronic respiratory diseases represent a substantial fraction, affecting hundreds of millions globally and contributing to millions of deaths each year emiological patterns confirm the scale of the problem but do not, by themselves, resolve its mechanistic complexity. Molecular investigations provide further clues. Cigarette smoke has long been validated as mutagenic, inducing oxidative DNA damage measurable through biomarkers such as 8-hydroxy-2′-deoxyguanosine (Valavanidis et al., 2009). Parallel studies demonstrate smoke-induced epigenetic remodeling, particularly alterations in histone acetylation that sustain pro-inflammatory gene expression and reduce therapeutic responsiveness (Rahman, 2005). Together, these findings suggest that smoke exposure does not merely injure cells transiently but reprograms them toward a stable, pathological state.
These observations point towards an unresolved challenge: cigarette smoke induced disease cannot be fully explained by isolated pathways or single-scale analyses. The information instead implies a tightly coupled system in which chemistry, cellular metabolism, immune response, behavior, and genetics interact nonlinearly. This realization naturally motivates a shift in direction from reductionist descriptions toward integrative, multi-scale models capable of capturing feedback, amplification, and emergent behavior.
Accordingly, this review adopts a independent research-driven perspective, assembling established findings across chemistry, cell biology, organ pathology, and systemic outcomes to identify where current explanations fall short. By treating cigarette smoke as a complex system perturbation rather than a discrete toxic exposure, the aim is to clarify mechanistic gaps and outline new conceptual directions for resolving its biological complexity. Such an approach is essential if future strategies are to move beyond damage cataloging toward predictive understanding and effective intervention.
2. The Exogenous Stressor: Physico-Chemical Characterization of Cigarette Smoke
The mechanisms underlying cigarette smoke–associated pathology must begin with a precise definition of the exposure itself. Cigarette smoke does not represent a singular toxic compound; rather, it is a chemically unstable aerosol generated through combustion and rapid cooling. It contains several thousand chemically diverse constituents that segregate into two interacting domains a particulate phase and a gaseous phase. Although chemically distinct, these domains operate in concert, forming a unified exposure system capable of delivering reactive molecules, long-lived radicals, and catalytic species within a single inhalation. This multiphase configuration is fundamental to the biological impact of cigarette smoke.
2.1 The Particulate Phase as a Sustained Redox Reservoir
The particulate component, commonly referred to as tar, functions as a condensed organic matrix enriched with chemically persistent toxic agents. It comprises polycyclic aromatic hydrocarbons, aromatic and heterocyclic amines, nitrosamines, and inorganic elements embedded within a lipophilic scaffold. Crucially, this matrix is not chemically passive. It stabilizes redox-active structures, particularly the quinone–hydroquinone semiquinone system. Semiquinone radicals within tar are maintained through resonance delocalization, allowing them to persist far longer than most combustion-generated radicals. Rather than being rapidly neutralized, these species retain the capacity for repeated electron-transfer reactions after deposition in aqueous biological environments. In addition, the particulate fraction accumulates transition metals, notably iron and copper. These metals remain largely inactive during aerosol transport but become catalytically engaged following pulmonary deposition, where they facilitate redox reactions that markedly intensify oxidative injury.
2.2 The Gaseous Phase as a Source of Acute Chemical Reactivity
In contrast to the relative persistence of tar-associated species, the gaseous phase represents the immediately reactive component of cigarette smoke. Each inhalation introduces extremely large numbers of transient radical species that react almost instantaneously with airway lining fluids. Among these constituents, nitric oxide is present at concentrations far exceeding physiological signaling ranges, shifting its role from regulatory mediator to a precursor of oxidative and nitrative stress. The gas phase also carries volatile aldehydes produced during combustion, including acrolein and formaldehyde. These electrophilic compounds readily form covalent bonds with nucleophilic sites on proteins, nucleic acids, and membrane lipids. Additional aldehydic species are generated secondarily when smoke-induced oxidation of endogenous lipids occurs, thereby extending chemical damage beyond the moment of exposure.
2.3 Electronic Properties and Radical Longevity
The biological behavior of cigarette smoke constituents is determined primarily by their electronic structure rather than by molecular classification. Gas-phase radicals possess unpaired electrons that confer extreme reactivity but confer only fleeting lifetimes, confining their effects to rapid reactions at epithelial surfaces. By contrast, the particulate phase contains paramagnetic radicals that persist long enough to penetrate cellular compartments and sustain redox activity over extended timeframes. This contrast establishes two distinct modes of injury: immediate surface oxidation followed by delayed, intracellular redox disruption.
2.4 Combustion Processes and Aerosol Formation
The chemical complexity of cigarette smoke originates directly from combustion dynamics. A burning cigarette acts as a microscale chemical reactor, reaching temperatures in excess of 800 °C. At these temperatures, radical-rich vapors are generated and subsequently cooled as they traverse the cigarette rod. During cooling, semi-volatile compounds condense onto forming particles, producing an aerosol dominated by fine particulate matter with aerodynamic diameters typically below 2.5 µm. This size range is both chemically and biologically significant. Fine particles evade mucociliary clearance mechanisms and deposit efficiently within alveolar regions, ensuring deep pulmonary delivery of tar-bound radicals and metals to areas characterized by high surface area and vulnerable cellular structures.
2.5 Coupled Redox Pathways and Catalytic Escalation
The chemical toxicity of cigarette smoke arises from interconnected reaction pathways rather than isolated molecular events. Upon hydration in lung fluids, stabilized semiquinone radicals transfer electrons to molecular oxygen, generating superoxide and converting a persistent radical into an active oxidant source. Superoxide subsequently undergoes dismutation to hydrogen peroxide, which functions as a diffusible intermediate. In the presence of iron or copper, hydrogen peroxide is converted into hydroxyl radicals through metal-catalyzed reactions. These radicals exhibit near-diffusion-limited reactivity and minimal selectivity, enabling indiscriminate damage to lipids, proteins, and nucleic acids. Further escalation occurs when superoxide reacts with nitric oxide to generate peroxynitrite, a highly reactive species capable of inducing both oxidative and nitrative modifications. This convergence demonstrates that the most destructive chemical species are not inhaled directly but are generated locally through synergistic redox chemistry involving both smoke phases.
2.6 Relevance within Inhalation Chemistry
The physicochemical characteristics outlined above distinguish combustible cigarette smoke from most other inhaled aerosols. While alternative exposures may introduce reactive aldehydes, they generally lack the particulate framework required to stabilize long-lived radicals and concentrate catalytic metals. The convergence of high radical density, sustained redox cycling, and efficient formation of secondary oxidants positions cigarette smoke among the most chemically aggressive inhalational stressors encountered by the respiratory system. This provides a reference model for evaluating oxidative potential and mechanistic complexity across inhalation toxicology.
3. The First Endogenous System: Fundamentals of Redox Biology
3.1 Reactive Species as Functional Regulators of Cellular Order
Cellular redox biology is structured around three interconnected classes of chemically reactive entities: oxygen, nitrogen, and sulfur-derived species (Lushchak, 2014). These molecules were once regarded as incidental by-products of aerobic metabolism, tolerated rather than utilized. This interpretation has been fundamentally revised. Contemporary evolutionary and cell biology now recognize reactive species as integral components of intracellular communication systems that contributed to the emergence of complex eukaryotic regulation (Checa & Aran, 2020).
The influence of these species follows a sharply defined concentration-dependent logic. When maintained within narrow physiological limits, they participate in regulated signaling processes a condition often described as oxidative eustress. In this cluster, relatively stable molecules such as hydrogen peroxide display the diffusion range and chemical selectivity required to modify specific redox-sensitive residues on target proteins, thereby supporting homeostasis and adaptive signaling (Circu & Aw, 2010; Sies, 2017). This balance is fragile, once oxidant production exceeds buffering capacity, signaling fidelity is lost. Initially reversible modifications give way to permanent chemical damage affecting lipids, proteins, and nucleic acids, marking the transition toward oxidative distress and chronic pathology (Dröge, 2002; Sies, 2017). Within this continuum, individual reactive species serve distinct roles. Nitric oxide, originally characterized as endothelium-derived relaxing factor, is essential for vascular regulation and pulmonary signaling under normal conditions (Palmer et al., 1987. Rapid combination with superoxide produces peroxynitrite, a potent oxidant that erases the functional boundary between signaling and toxicity (Pacher et al., 2007; Radi, 2018). Superoxide itself represents the first radical generated during oxygen reduction and is tightly controlled by superoxide dismutase, whose identification established the enzymatic foundation of antioxidant defense (McCord & Fridovich, 1969).
3.2 Intrinsic Sources of Reactive Species
The most significant of these is the mitochondrial electron transport chain. Mitochondria are not simply energy-producing organelles; they are continuous sources of redox activity arising from the unavoidable inefficiencies of aerobic respiration (Shadel & Horvath, 2015). Within mitochondria, superoxide production arises through two mechanistically distinct routes. Forward electron transfer accompanies normal respiration and produces low-level reactive species as a background signal. Reverse electron transfer, in contrast, occurs under conditions of elevated membrane potential or metabolic imbalance and can generate intense bursts of reactive species (Forrester et al., 2018). These properties position mitochondria as both detectors and amplifiers of metabolic stress.
Distinct from mitochondrial leakage, the NADPH oxidase family produces reactive species intentionally. These enzymes generate superoxide in a regulated manner for immune defense and signal transduction (Finkel, 2011). Among them, NOX4 occupies a unique position through its mitochondrial localization, providing a direct interface between receptor-driven signaling and mitochondrial redox output (Forrester et al., 2018). Additional contributors include peroxisomes, xanthine oxidase, enzymes of the arachidonic acid cascade, and alpha-keto acid dehydrogenase complexes. Under certain metabolic conditions, complexes such as KGDHC and PDC generate reactive species at rates exceeding those of mitochondrial complex I (Forrester et al., 2018). Even normal physiological states, such as physical exercise, transiently elevate oxidative load a response that, when constrained, promotes adaptive remodeling rather than injury (Powers & Jackson, 2008).
3.3 Radical Reactivity Through the Lens of Chemical Physics
The interaction between smoke-derived oxidants and cellular macromolecules is governed by fundamental physical laws rather than stochastic chemistry. Biological reactions proceed along defined potential energy surfaces, transitioning through activated states rather than occurring randomly (Pauling, 1960; Marcus, 1993). Reaction likelihood is determined by frontier molecular orbital compatibility, where electron transfer depends on the energetic alignment of donor and acceptor orbitals (Fukui, 1982). These reactions unfold within an aqueous environment that actively shapes chemical outcomes. Water facilitates proton exchange, stabilizes transient intermediates, and enables redox cycling, functioning as a reactive participant rather than an inert solvent (Ball, 2017). Within this matrix, transition metals such as iron and copper catalyze the formation of hydroxyl radicals via Fenton chemistry, producing species characterized by extreme reactivity and minimal selectivity (Halliwell & Gutteridge, 1984). When hydroxyl radicals attack lipid bilayers, they initiate chain reactions of lipid peroxidation that propagate independently of the initiating event. These cascades alter membrane structure and generate secondary aldehydes with prolonged biological activity (Esterbauer et al., 1991; Gaschler & Stockwell, 2017). Such reactions threaten homochirality the asymmetric molecular organization essential for enzymatic specificity and biological order which emerged early in chemical evolution and remains a defining feature of life (Blackmond, 2010).
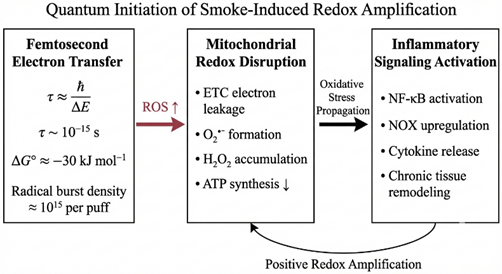
Figure 3.3.1 Femtosecond-scale electron transfer (τ ~ 10?¹? s) generates a high radical flux (~10¹? per puff; ΔG° ≈ −30 kJ mol?¹), inducing mitochondrial ETC leakage, ROS amplification, ATP decline, and sustained inflammatory signaling via a positive redox feedback loop.
3.4 Redox Couples as Regulatory Networks
To persist under constant oxidative challenge, cells rely on interconnected redox buffering systems rather than isolated scavengers. The glutathione, thioredoxin, and pyridine nucleotide couples form a dynamic network that maintains redox balance while enabling controlled signaling (Circu & Aw, 2010; Ray et al., 2012). These systems regulate protein function through reversible oxidation of cysteine residues. This mechanism commonly referred to as the cysteine switch allows reactive species to induce transient structural and functional changes in proteins via sulfenic acid formation. The process is functionally analogous to phosphorylation, enabling redox-based regulation to integrate seamlessly with established signaling pathways (Finkel, 2011; Holmström & Finkel, 2014).
3.5 Spatial Redox Control and the Origins of Pathology
Redox regulation is not uniform throughout the cell. Distinct compartments maintain specialized redox environments, creating gradients that guide processes such as growth factor signaling, immune activation, and resolution of inflammation (Finkel, 2011). Disease emerges when these gradients are disrupted. Excessive oxidant production or failure of buffering systems conditions exacerbated by cigarette smoke collapse compartmentalized control, allowing localized signaling events to escalate into widespread molecular damage. This loss of spatial regulation underlies the shared redox basis of diverse disorders, including cardiovascular disease, metabolic dysfunction, and neurodegeneration (Dröge, 2002; He & Zuo, 2020). Assessment of this balance has historically relied on stable radical assays, such as DPPH-based methods, which continue to provide reference benchmarks for oxidative capacity and therapeutic efficacy (Blois, 1958). Collectively, these observations support a central conclusion: pathology does not arise from reactive species themselves, but from the breakdown of the systems.
4. The Second Endogenous System: The ATP Bioenergetic and Signaling Axis
4.1 From Quantum to Molecule: The Energetics of ATP
While the redox system maintains the electronic integrity of the cell, the bioenergetic system sustains its thermodynamic viability. At the center of this system lies Adenosine Triphosphate (ATP), a molecule that transcends its role as a mere chemical compound to function as the universal currency of biological work. To understand the pathogenic collapse induced by cigarette smoke, one must first appreciate the quantum mechanical precariousness of the energy source it depletes. The bioenergetic potency of ATP acts according to the thermodynamic instability of its phosphoanhydride bonds. Often colloquially termed "high-energy bonds". In reality, the potential energy resides not in the bond itself, but in the substantial electrostatic repulsion between the negatively charged oxygen atoms of the phosphate groups. Upon hydrolysis, this repulsion is relieved, releasing a significant quantity of Gibbs free energy (δ-G approx -30.5 kJ/mol) that drives unfavorable biological reactions. This molecular tension makes ATP an inherently metastable molecule essential for rapid energy transfer but exquisitely sensitive to metabolic disruption.
4.2 Intracellular Energy Currency
The synthesis of this currency is the primary mandate of cellular metabolism, achieved through two distinct but integrated pathways: cytosolic glycolysis and mitochondrial oxidative phosphorylation. While glycolysis provides rapid, anaerobic ATP generation, the bulk of cellular energy is derived from the mitochondrion via the mechanism of chemiosmotic coupling. First elucidated by Peter Mitchell, this theory posits that the energy from electron transfer is stored as an electrochemical proton gradient (δ-psi/m) across the inner mitochondrial membrane, which subsequently drives the ATP synthase rotary motor (Mitchell, 1961). This process is the foundational principle of bioenergetics; however, it is also the system's Achilles' heel, as the electron transport chain required to generate this gradient is the primary site of ROS leakage. The cell monitors its bioenergetic health through a precise homeostatic sensor known as the "Energy Charge." This metric is defined not by the absolute concentration of ATP, but by the ratio of ATP to its hydrolysis products, ADP and AMP. Under physiological conditions, this ratio is tightly conserved. However, metabolic stress such as that induced by the toxic constituents of cigarette smoke precipitates a drop in this ratio. This bioenergetic deficit triggers the activation of AMP-activated protein kinase (AMPK), signaling a desperate shift from anabolic growth to catabolic survival, a state that often precedes apoptotic cell death.
4.3 Extracellular Signaling: eATP as a DAMP
A paradigm shift in our understanding of bioenergetics occurred with the realization that ATP functions as a "Dr. Jekyll and Mr. Hyde" molecule. While intracellular ATP is the currency of life, extracellular ATP (eATP) is a harbinger of danger. In healthy tissues, ATP is confined within the cell. However, during events of necrotic lysis or controlled efflux via Pannexin/Connexin channels events triggered by cigarette smoke exposure ATP is released into the extracellular milieu. Once outside the cell, ATP transforms into a potent Damage-Associated Molecular Pattern (DAMP). It binds to the Purinergic Receptor network, a widespread family of cell-surface receptors divided into the P2X (ionotropic) and P2Y (metabotropic) classes. Of particular relevance to this review is the P2X7 receptor. When activated by high concentrations of eATP, the P2X7 receptor initiates a signaling cascade that bridges bioenergetics and inflammation, acting as a key trigger for the assembly of the NLRP3 inflammasome. Thus, the leakage of the cell's fuel becomes the spark for systemic inflammation.
4.4 The ATP-Redox Dysregulation: A Critical Interdependence
This interdependence is governed by the principle that metabolic flux determines redox state, and conversely, redox state regulates metabolic flux (Hayes & Dinkova-Kostova, 2014). This operates nexus as a bidirectional feedback loop. First, bioenergetic collapse drives oxidative stress; as mitochondrial function fails and the proton gradient dissipates, the electron transport chain becomes highly reduced, dramatically increasing the probability of electron slippage and superoxide formation. Conversely, oxidative stress drives bioenergetic collapse; the ROS generated by cigarette smoke directly target and inactivate the iron-sulfur clusters of the electron transport chain enzymes and the ATP synthase itself. This creates a pathogenic spiral: smoke damages the mitochondrial engine, causing it to leak toxic radicals, which in turn further damage the engine, leading to a catastrophic failure of both energy production and redox homeostasis.
5. Endogenous Antioxidant Defenses as a Systems-Level Countermeasure
5.1 Low–Molecular Weight Antioxidants: Immediate Chemical Buffering
To withstand sustained oxidative challenge from environmental stressors, biological systems rely on a layered defense architecture. The most immediate layer consists of small, diffusible antioxidant molecules that neutralize reactive intermediates through direct chemical sacrifice. Among these, glutathione occupies a central position. Present at millimolar concentrations in the cytosol, this tripeptide does far more than intercept radicals. It establishes and preserves the reducing environment required for nucleotide synthesis, protein folding, and DNA repair, functioning as the principal intracellular redox currency. This thiol-based buffering system is reinforced by dietary antioxidants with complementary physicochemical niches. Ascorbate operates predominantly within aqueous compartments, while tocopherols protect lipid membranes from peroxidative chain reactions. Despite their importance, these molecules are constrained by stoichiometry: each antioxidant molecule can neutralize only a finite number of radicals. When reactive species are generated catalytically as occurs during sustained smoke exposure non-enzymatic scavengers alone are rapidly overwhelmed, necessitating enzymatic amplification.
5.2 Enzymatic Antioxidants: Catalysis Over Stoichiometry
The second defensive tier resolves this limitation through catalysis. Enzymatic antioxidants are capable of processing vast quantities of reactive species without being consumed, providing scalable protection against oxidative escalation. At the foundation of this system lies superoxide dismutase, whose discovery established that antioxidant defense is not merely chemical but enzymatic in nature (McCord & Fridovich, 1969). Superoxide dismutase rapidly converts superoxide anion into hydrogen peroxide, preventing its participation in reactions that generate peroxynitrite. However, hydrogen peroxide itself remains chemically active. Its regulation is therefore delegated to downstream enzymes, most notably catalase and the glutathione peroxidase family. These enzymes operate in coordination with peroxiredoxins and the thioredoxin system, maintaining peroxide concentrations within a narrow range that permits signaling while preventing accumulation sufficient to cause cellular injury (Ryter et al., 2007; Barouki & Morel, 2011). This enzymatic network functions not as a blunt detoxification tool, but as a finely tuned modulator of redox tone.
5.3 Nrf2 as the Central Genomic Integrator of Antioxidant Defense
While constitutive enzymes and scavengers provide baseline protection, adaptation to intense or prolonged stress requires transcriptional reprogramming. This adaptive response is orchestrated by Nuclear Factor Erythroid 2–Related Factor 2 (Nrf2), which functions as the principal genomic regulator of antioxidant and detoxification pathways (Baird & Dinkova-Kostova, 2011; Ryter et al., 2007). Initially identified in the mid-1990s as a member of the Cap-n-Collar transcription factor family (Moi et al., 1994), Nrf2 has since emerged as the core cytoprotective response to electrophilic and oxidative insults (Kensler et al., 2007). Under resting conditions, Nrf2 is continuously synthesized but remains functionally silent due to sequestration by its cytoplasmic repressor Keap1. Acting as a redox-sensitive adaptor for a Cul3-based ubiquitin ligase complex, Keap1 ensures rapid Nrf2 degradation, preventing unnecessary gene expression (Zhang et al., 2003). Oxidative or electrophilic stress alters this equilibrium. Modification of key cysteine residues within Keap1 induces conformational changes that disrupt its ability to target Nrf2 for degradation. This releases Nrf2, allowing it to accumulate and translocate to the nucleus, where it dimerizes with small Maf proteins and binds antioxidant response elements in target gene promoters (Itoh et al., 1997). The resulting transcriptional program induces a broad spectrum of Phase II enzymes, including heme oxygenase-1 and NAD(P)Hydroquinone oxidoreductase-1, expanding cellular capacity for detoxification and redox control.
5.4 Network Integration with Inflammatory, Mitochondrial, and Xenobiotic Pathways
Nrf2 does not function as an isolated switch but as a node within a densely interconnected signaling network. Its activity intersects with NF-κB signaling, influencing the balance between cytoprotection and inflammatory activation (Barouki & Morel, 2011). Beyond canonical regulation, additional pathways link Nrf2 activation to cellular quality control mechanisms. The autophagy adaptor p62 provides one such link. By competing with Nrf2 for Keap1 binding, p62 stabilizes Nrf2 during conditions of proteotoxic stress, coupling antioxidant gene induction to autophagic flux (Komatsu et al., 2010). Mitochondrial signaling further integrates into this system through proteins such as PGAM5, which associate with the Keap1-Nrf2 complex and convey mitochondrial stress signals directly to nuclear transcriptional responses (Lo & Hannink, 2006). In parallel, the aryl hydrocarbon receptor activated by polycyclic aromatic hydrocar present in cigarette smoke interacts with Nrf2-regulated pathways, creating an adaptive axis specifically tuned for xenobiotic metabolism (Rojo de la Vega et al., 2016).
5.5 Redox Hormesis and the Dual Nature of Nrf2 Activation
The protective capacity of the antioxidant system is governed by hormesis rather than linear benefit. Transient activation of Nrf2 enhances cellular resilience and mitigates smoke-induced injury (Kensler et al., 2007). However, persistent or uncontrolled activation produces adverse outcomes. In multiple malignancies, particularly lung adenocarcinoma, somatic mutations in KEAP1 lead to constitutive Nrf2 signaling. In this pathological context, Nrf2 no longer serves a protective role. Instead, it shields malignant cells from oxidative damage, suppresses apoptotic signaling, and supports metabolic rewiring that favors rapid proliferation (Sporn & Liby, 2012; Taguchi & Yamamoto, 2017). Thus, the same system that preserves normal tissue integrity under stress becomes a driver of therapeutic resistance and disease progression when dysregulated. Taken together, endogenous antioxidant defenses represent not a static shield but a dynamic regulatory system. Their effectiveness depends on precise control of magnitude, duration, and spatial deployment conditions that are profoundly disrupted by chronic smoke exposure.
6. Mechanistic Toxicology: The Interface of Smoke and Cell
6.1 From Chemical Exposure to Biological Injury
Constitute with the chemical characteristics of cigarette smoke and the intrinsic redox architecture now defined, the analysis turns to the point of convergence where exogenous chemistry intersects living tissue. This interface represents the earliest mechanistic conversion of exposure into injury the molecular “first strike” that initiates disease progression. Importantly, smoke-induced pathology does not unfold as a simple cause effect sequence. Instead, it evolves through a self-reinforcing feedback loop commonly described as the inflammation oxidation cycle (Boots et al., 2009).This cycle is rooted in a fundamental toxicological imbalance. When the extreme oxidant burden delivered by cigarette smoke on the order of 10¹? reactive species per puff-penetrates the epithelial lining fluid, it overwhelms immediate chemical defenses such as glutathione and ascorbate. Once these front-line buffers are exhausted, reactive species gain direct access to membrane lipids and surface proteins of airway epithelial cells. At this point, the effective dose is no longer defined solely by the inhaled smoke, but by the secondary reactive species generated endogenously by injured tissue and recruited inflammatory cells (Rahman, 2005).

Figure 6.1.1 | Smoke-induced reactive species drive redox imbalance, mitochondrial dysfunction, inflammatory amplification and genomic instability, culminating in organ-level pathology.
6.2 Redox-Driven Protease Dysregulation and Structural Lung Damage
One of the earliest and most consequential outcomes of this oxidative shift is disruption of protease control. Under physiological conditions, proteolytic enzymes released by inflammatory cells particularly neutrophil elastase are tightly regulated by endogenous inhibitors. Chief among these is α?-antitrypsin, which neutralizes elastase activity and preserves alveolar integrity. This balance is exquisitely sensitive to oxidation. The reactive center of α?-antitrypsin contains a methionine residue whose oxidation state determines inhibitory function. Reactive oxygen and nitrogen species abundant in cigarette smoke, especially peroxynitrite, selectively oxidize this residue to methionine sulfoxide. This seemingly minor chemical modification abolishes the inhibitor’s ability to bind elastase (Rahman, 2005). The consequences are structural rather than subtle. Unchecked elastase activity degrades elastin fibers within the alveolar septa, progressively eroding the architectural framework of the lung. This oxidation-driven inactivation of antiproteases constitutes the central molecular mechanism underlying emphysema, linking a discrete chemical reaction to irreversible organ-level destruction (Boots et al., 2009).
6.3 Electrophilic Stress: Carbonyl Chemistry, Quinones, and Redox Collapse
While radical species produce immediate oxidative injury, longer-lasting toxicity arises from electrophilic constituents embedded within the particulate phase. Reactive aldehydes such as acrolein and 4-hydroxynonenal act as soft electrophiles, selectively targeting nucleophilic thiol groups on cysteine residues through Michael addition reactions (Esterbauer et al., 1991). The primary intracellular sink for these reactions is glutathione. Covalent conjugation between aldehydes and glutathione rapidly depletes the cellular thiol pool, undermining antioxidant capacity at its foundation (Givi et al., 2020). This depletion has dual consequences: it compromises direct radical scavenging and disrupts redox-sensitive signaling pathways that depend on glutathione availability. Quinones introduced with tar compounds further destabilize the intracellular environment through redox cycling. After enzymatic reduction to hydroquinones, these molecules are readily re-oxidized by molecular oxygen, regenerating superoxide in a futile catalytic loop. This process effectively converts the cell interior into a persistent source of reactive oxygen species, sustaining oxidative stress long after external exposure has ended (Givi et al., 2020).
6.4 Metabolic Activation of PAHs and Genetic Injury
Polycyclic aromatic hydrocarbons exemplify a distinct mode of action as pro-carcinogens requiring metabolic activation. Upon cellular entry, these hydrophobic molecules bind the aryl hydrocarbon receptor, initiating nuclear translocation and transcriptional induction of cytochrome P450 enzymes, particularly CYP1A1 and CYP1B1 (Rojo de la Vega et al., 2016). This response reflects an attempted detoxification. However, the enzymatic metabolism of PAHs produces highly reactive diol-epoxide intermediates. These electrophiles intercalate into DNA and form covalent adducts with guanine bases, distorting the DNA helix and interfering with replication fidelity. The resulting mispairing events characteristically G→T transversions represent the molecular fingerprint of tobacco-induced mutagenesis, a phenomenon classically demonstrated by the Ames mutagenicity assay (Ames et al., 1973; Boots et al., 2009).
6.5 Metal-Catalyzed Radical Amplification
The severity of all preceding mechanisms is magnified by the presence of transition metals deposited with cigarette smoke. Iron and copper accumulate within the epithelial lining fluid, where they interact with hydrogen peroxide generated through enzymatic dismutation of superoxide. These metals catalyze Fenton-type reactions, converting hydrogen peroxide into hydroxyl radicals species characterized by extreme reactivity and virtually no molecular selectivity (Halliwell & Gutteridge, 1984). Hydroxyl radicals react at diffusion-controlled rates with nearby biomolecules, ensuring immediate damage to DNA, lipids, or proteins. This catalytic amplification loop ensures that even modest oxidative inputs are translated into disproportionate biological injury. The process illustrates a central principle of mechanistic toxicology: the pathological impact of cigarette smoke arises not from individual constituents in isolation, but from their capacity to interact synergistically within biological systems, producing outcomes far more destructive than the sum of their chemical parts.
7. Molecular and Genetic Regulation: The Corruption of the Blueprint
7.1 DNA Damage and Repair: Permanent Molecular Imprints
The chemical injury described earlier ultimately converge on the cell’s most critical asset: genomic integrity. DNA stability, as first clarified through its double-helical architecture, depends on precise base pairing and controlled electronic interactions. Oxidative stress undermines this stability not by indiscriminate fragmentation, but by introducing chemically defined lesions that compromise replication fidelity. Among these, oxidation of guanine produces 8-hydroxy-2′-deoxyguanosine, the most abundant and biologically consequential oxidative DNA lesion. Guanine’s electron-rich structure makes it a preferred target for hydroxyl radicals, and once modified, it mispairs during replication, driving G:C to T:A transversions (Valavanidis et al., 2009). This mutation pattern is not stochastic; it represents a reproducible molecular fingerprint of oxidative mutagenesis that mechanistically links radical chemistry to carcinogenic transformation (Ames et al., 1973).
Cells counter these lesions through coordinated DNA damage response systems, primarily base excision repair and nucleotide excision repair. Emerging evidence indicates that these pathways are not diffusely organized but instead assemble into transient repair hubs through liquid–liquid phase separation. These condensates concentrate repair enzymes at damage sites, increasing efficiency but introducing a new vulnerability: their formation and stability depend on finely balanced electrostatic and redox conditions. In the oxidatively burdened environment characteristic of smoke-exposed lung tissue, this biophysical organization becomes destabilized, impairing repair efficiency despite intact enzymatic machinery (Jodeiri Akbar et al., 2023).
7.2 RNA Oxidation and Loss of Translational Precision
DNA damage has long dominated toxicological narratives, yet RNA oxidation represents an earlier and often underestimated point of failure. Unlike DNA, RNA lacks histone protection and is predominantly single-stranded, rendering it highly accessible to reactive species within the cytosol. Oxidative modification of messenger RNA alters codon integrity and disrupts ribosomal progression. This process produces translational errors that generate truncated or improperly folded polypeptides. The resulting defective proteins enter the cytoplasm before quality-control systems can intervene, placing immediate strain on proteostasis networks. In this way, oxidative RNA damage seeds cellular dysfunction upstream of protein maturation, introducing faulty molecular components that propagate stress throughout the intracellular environment (Boots et al., 2009).
7.3 Epigenetic Reprogramming as a Long-Term Stress Record
Beyond direct nucleic acid damage, cigarette smoke induces durable changes in gene expression without altering DNA sequence. This epigenetic reprogramming arises from redox-dependent modulation of chromatin-modifying enzymes. Nuclear oxidative stress directly impairs the activity of histone deacetylases and acetyltransferases, disrupting the dynamic equilibrium that governs chromatin accessibility. A particularly critical target is HDAC2. Oxidative inhibition of this enzyme prevents removal of acetyl groups from histones at inflammatory gene promoters, maintaining chromatin in a transcriptionally permissive state. The consequence is sustained overexpression of pro-inflammatory mediators such as IL-8, independent of ongoing exposure. Clinically, this mechanism explains a central paradox in chronic obstructive pulmonary disease: resistance to corticosteroid therapy. Steroids require functional HDAC2 to repress inflammatory gene expression; when this epigenetic regulator is oxidatively disabled, pharmacological intervention loses efficacy (Rahman, 2005).
7.4 Collapse of Regulatory Signaling Networks
The cumulative effect of genetic lesions, translational errors, and epigenetic fixation is a breakdown of the cell’s regulatory hierarchy. This breakdown displays as dysregulated transcriptional signaling, conceptualized as competition among dominant control pathways. Inflammatory amplification is driven primarily by nuclear factor-κB. Under oxidative stress, inhibitory constraints on this transcription factor are chemically disrupted, allowing rapid nuclear accumulation. Once active, NF-κB induces expression of cytokines, chemokines, and adhesion molecules, converting epithelial cells into active participants in immune recruitment and perpetuating local inflammation (Rahman, 2005).
Counterbalancing this response is the antioxidant transcription factor Nrf2. Under physiological conditions, Nrf2 activity restrains inflammatory escalation through coordinated cytoprotective gene expression. Chronic smoke exposure, however, overwhelms this axis. Sustained oxidative pressure leads to functional exhaustion or dysregulation of Nrf2 signaling, allowing inflammatory drivers to dominate unchecked (Ryter et al., 2007). Simultaneously, escalating DNA damage engages p53-mediated surveillance pathways. This engagement forces a binary cellular decision governed by mitogen-activated protein kinase signaling: transient cell-cycle arrest to permit repair, or initiation of apoptosis once damage surpasses repair capacity. Biomarkers such as accumulated 8-OHdG reflect this tipping point. In carcinogenic contexts, mutational inactivation of p53 disables this safeguard, permitting survival and proliferation of genetically compromised cells. This failure marks the transition from chronic inflammation to malignant transformation, completing the mechanistic arc from chemical exposure to cancer development.
8. Cellular Entry and Early Structural Failure Under Smoke Exposure
8.1 Airway Epithelium as an Active Gatekeeper
Smoke-induced pathology originates at the anatomical boundary separating the external environment from internal physiology: the airway epithelium. Far from functioning as an inert sieve, this tissue operates as a highly regulated barrier composed of ciliated epithelial cells, mucus-secreting goblet cells, and basal progenitor populations. Its integrity is maintained through intercellular junctional complexes that tightly regulate permeability. The principal route of entry for soluble smoke constituents arises through disruption of this junctional architecture. Exposure to fine particulate matter (PM?.?) and reactive gases destabilizes tight junction and adhere junction assemblies, selectively compromising proteins such as zonula occludens-1 and E-cadherin. The functional consequence is a measurable reduction in trans-epithelial electrical resistance, reflecting loss of barrier cohesion. This phenomenon barrier dysfunction constitutes a core pathogenic event in both asthma and chronic obstructive pulmonary disease, as it permits accelerated diffusion of toxicants into sub-epithelial compartments and amplifies immune activation (Nakayama et al., 2017). Experimental dissection of this process relies on well-defined epithelial models. Primary cultures of normal human bronchial epithelial cells, propagated under serum-free conditions, provide a physiologically relevant platform for studying barrier responses, a methodology established in the early 1980s (Lechner et al., 1982). These systems are complemented by the A549 alveolar epithelial line, widely employed to examine toxin transport, stress signaling, and carcinogenic transformation within distal lung epithelium (Foster et al., 1998).
8.2 Ultrafast Intracellular Responses: The Kinetic Phase of Injury
Following epithelial breach, intracellular damage unfolds on a rapid kinetic timescale. The earliest events occur within milliseconds to minutes and are dominated by direct chemical modification of proteins and membranes. Highly reactive oxidants penetrate the cytosol and immediately engage the redox-sensitive proteome. Cysteine thiol groups represent primary targets, undergoing oxidation to sulfenic acid derivatives or forming aberrant disulfide bonds that alter protein conformation and activity. Concurrently, oxidative attack extends to the protein backbone through carbonylation reactions. Side chains of proline, arginine, lysine, and threonine are converted into aldehyde or ketone groups, introducing irreversible structural damage. Unlike reversible thiol oxidation, protein carbonylation promotes unfolding and aggregation, overwhelming degradation systems. For this reason, total protein carbonyl content remains one of the most robust biochemical indicators of severe oxidative stress (Dalle-Donne et al., 2003). Membrane integrity is compromised in parallel through lipid peroxidation. Reactive species initiate chain reactions within polyunsaturated fatty acids of the phospholipid bilayer, progressively altering membrane fluidity and permeability. These changes violate the principles of the fluid mosaic organization of membranes and generate diffusible aldehydes such as 4-hydroxynonenal. These secondary products migrate away from the membrane to form covalent adducts with cytosolic proteins, extending the zone of damage beyond the initial site of oxidation (Gaschler & Stockwell, 2017).
8.3 Organelle-Specific Vulnerability and Convergent Failure
Smoke-induced cytotoxicity is spatially organized rather than uniform. Individual organelles exhibit distinct susceptibilities, and damage within these compartments activates specialized stress programs that ultimately converge on irreversible cell fate decisions.
8.3.1 Mitochondria: Energy Failure and Defective Quality Control
Mitochondria occupy a paradoxical position as both generators and casualties of oxidative stress. Exposure to cigarette smoke extract rapidly dissipates the mitochondrial membrane potential, arresting oxidative phosphorylation and precipitating abrupt ATP depletion. Under conditions of extreme redox stress, the mitochondrial permeability transition pore opens, allowing solute influx, matrix swelling, and rupture of the outer membrane. This rupture releases cytochrome c into the cytosol, activating the caspase cascade that executes apoptotic cell death, a process formally described in the early 1970s (Kerr et al., 1972). To counteract this threat, cells initiate mitophagy, selectively isolating dysfunctional mitochondria for lysosomal degradation. However, chronic smoke exposure impairs mitophagic efficiency, permitting accumulation of damaged mitochondria that persist as continuous sources of reactive oxygen species functionally inert yet metabolically hazardous structures (Ryter & Choi, 2016; Shadel & Horvath, 2015).
8.3.2 Endoplasmic Reticulum: Adaptive Folding to Terminal Stress
Oxidative modification of nascent and mature proteins places substantial strain on the endoplasmic reticulum, the central hub of protein folding. Accumulation of misfolded proteins activates the unfolded protein response, initially enhancing chaperone expression and attenuating protein synthesis in an effort to restore equilibrium. Under sustained smoke exposure, this adaptive program fails. Prolonged stress signaling converts the unfolded protein response into a pro-apoptotic pathway, marking the collapse of proteostasis. In this state, the endoplasmic reticulum ceases to function as a biosynthetic organelle and instead becomes an active participant in cell death signaling (Hetz, 2012).
8.3.3 Lysosomes and Autophagy: Defensive Recycling Versus Catastrophic Leakage
Lysosomes, essential for intracellular recycling, are particularly sensitive to oxidative damage. Reactive species destabilize lysosomal membranes, leading to lysosomal membrane permeabilization and uncontrolled release of cathepsins into the cytosol. This event promotes widespread macromolecular degradation and necrotic cell death. Paradoxically, lysosomes also coordinate autophagy, a critical survival mechanism during stress (Mizushima et al., 2008). Through a key signaling intersection, the autophagy adaptor p62 interferes with Keap1-mediated repression of Nrf2, thereby linking degradation of damaged components to activation of antioxidant gene expression (Komatsu et al., 2010). Chronic smoke exposure frequently disrupts this coupling, severing communication between recycling and defense and leaving cells unable to adapt.
8.3.4 Structural Breakdown of the Plasma Membrane and Cytoskeleton
The final layer of damage targets cellular architecture itself. Cytoskeletal elements actin filaments and microtubules are highly responsive to redox conditions. Oxidative modification of these proteins leads to depolymerization, loss of structural tension, and impaired ciliary motion. These changes contribute directly to mucociliary dysfunction characteristic of chronic bronchitis. Simultaneously, lipid peroxidation compromises plasma membrane integrity, increasing calcium permeability and disrupting ionic homeostasis. The combined effect is a cell that is mechanically rigid, metabolically compromised, and incapable of maintaining barrier or transport functions. This condition favors necrotic death and release of inflammatory signals, perpetuating tissue-level damage and chronic inflammation.
9. Immune and Inflammatory Interference: The Bridge to Systemic Disease
9.1 Redox Control of Immune Cell Behavior
The molecular and structural injuries described in earlier chapters do not remain confined to epithelial cells. They function as distress signals that actively recruit and reprogram the immune system. This response is not reflexive but conditional, governed by the redox state of immune cells themselves. Both innate and adaptive immunity depend on finely tuned redox signaling for functional competence. The innate immune system, organized around pattern-recognition rather than clonal specificity, employs reactive oxygen species not only as antimicrobial weapons but also as signaling intermediates that regulate activation thresholds. A similar dependence exists within adaptive immunity. T-cell activation follows a kinetic proofreading framework in which the duration and stability of antigen–receptor interactions determine downstream signaling outcomes. Oxidative stress perturbs this timing mechanism by modifying phosphorylation dynamics within the T-cell receptor cascade, thereby degrading the cell’s ability to distinguish self from non-self.
This dysregulation converges on nuclear factor-κB, a transcription factor that occupies a central position in inflammatory control. Under basal conditions, NF-κB is restrained in the cytoplasm by inhibitory proteins. Smoke-derived reactive species promote phosphorylation and degradation of these inhibitors, enforcing persistent nuclear localization of NF-κB. The result is immune cells locked into a state of continuous activation, transforming protective inflammation into a driver of chronic tissue damage (Rahman, 2005).
9.2 Inflammasome Assembly as a Redox-Sensitive Signaling Hub
At the core of sterile inflammation lies the inflammasome, a multiprotein signaling platform that converts cellular stress into cytokine maturation. Among inflammasome subtypes, NLRP3 serves as the dominant sensor of smoke-induced damage signals. Activation of this complex requires two coordinated inputs. The first is a priming signal, in which redox-dependent NF-κB activation increases transcription of NLRP3 and pro-interleukin-1β. The second signal initiates physical assembly of the inflammasome and is provided by danger-associated molecular patterns generated during cellular injury. Extracellular ATP released from damaged cells engages purinergic receptors, inducing potassium efflux, while mitochondrial reactive oxygen species act as direct modulators of NLRP3 activation.
Once assembled, the inflammasome activates caspase-1, which cleaves pro-interleukin-1β into its mature, bioactive form. Interleukin-1β then propagates inflammation locally and systemically. Experimental studies in human bronchial epithelial systems demonstrate that selective interruption of this pathway using naturally derived compounds can halt inflammatory amplification, underscoring the inflammasome as a critical therapeutic node (Li et al., 2024).
9.3 Cytokine–ROS Feedback Loops and Inflammatory Persistence
Inflammatory cytokines do not represent terminal signaling products; they function as amplifiers of oxidative stress. Once interleukin-1β is released, it initiates a feed-forward loop in which macrophages and epithelial cells produce additional cytokines such as tumor necrosis factor-α and interleukin-8. These mediators stimulate NADPH oxidase activity in neutrophils and epithelial cells, driving a secondary wave of superoxide generation. This endogenous oxidative surge persists independently of continued smoke exposure, sustaining inflammation long after the initiating insult has ceased. In this way, oxidative stress and cytokine signaling become mutually reinforcing, creating a self-maintaining inflammatory phenotype that defines chronic lung disease (Boots et al., 2009).
9.4 Immunometabolism and Mitochondrial Signaling Integration
Recent advances in immunology have revealed that immune activation is inseparable from metabolic state. This concept, termed immunometabolism, is particularly relevant in smoke-exposed lungs. Alveolar macrophages undergo metabolic reprogramming in which classical energy-producing pathways are repurposed for signaling. Within this context, accumulation of mitochondrial metabolites such as succinate stabilizes hypoxia-inducible transcription factors, promoting interleukin-1β production. Mitochondrial reactive oxygen species generated during this metabolic shift are not incidental; they are required for antimicrobial defense. However, in the absence of infection, persistent metabolic activation produces sterile inflammation. This process intersects with antioxidant regulation. Crosstalk between redox-responsive transcriptional networks and intermediary metabolism determines whether metabolic reprogramming resolves injury or perpetuates damage. Under chronic smoke exposure, this balance collapses, converting an adaptive immune strategy into a pathological state (Hayes & Dinkova-Kostova, 2014; Schumacker, 2015).
9.5 Chronic Inflammation: Driver of Autoimmunity and Malignancy
Sustained inflammatory signaling fulfills a central criterion for malignant transformation. Persistent production of reactive species and cytokines generates a mutagenic tissue environment that promotes genomic instability and tumor initiation. A critical outcome of this inflammatory milieu is ferroptosis, an iron-dependent form of regulated cell death driven by unchecked lipid peroxidation. Unlike apoptosis, ferroptosis releases damage-associated signals that further activate immune pathways, intensifying tissue inflammation. In established tumors, particularly lung carcinoma models, mutations frequently lock antioxidant regulatory pathways into a constitutively active state. This adaptation shields malignant cells from ferroptotic death and therapeutic oxidative stress, conferring resistance to chemotherapy and radiotherapy (Sporn & Liby, 2012; Taguchi & Yamamoto, 2017).
Importantly, the consequences of this immune dysregulation extend beyond the lung. Bidirectional communication between the respiratory tract and the gastrointestinal system the gut–lung axis links pulmonary inflammation to microbial imbalance. Systemic inflammatory signals and swallowed particulates alter intestinal microbiota composition, generating metabolites that feed back to exacerbate lung pathology. This interorgan handshake illustrates that chronic obstructive lung disease represents a systemic failure of mucosal immunity rather than an isolated pulmonary disorder (Budden et al., 2017).
10. Systemic and Organ-Level Symptoms : The Clinical Outcome
10.1 The Respiratory System as the Primary Site of Conflict
The lung represents the principal point of contact between cigarette smoke and human biology, owing to its vast surface area and continuous exposure to the external environment. The molecular insults outlined for oxidative overload, proteolytic imbalance, and epithelial barrier failure coalesce clinically into Chronic Obstructive Pulmonary Disease. As articulated by contemporary consensus frameworks, COPD is not a single disorder but a spectrum of overlapping pathological states defined by persistent airflow limitation and progressive respiratory decline (Agustí et al., 2023). Disease progression follows two dominant and mechanistically distinct trajectories. The first involves parenchymal destruction. Oxidative modification of α?-antitrypsin eliminates its capacity to restrain neutrophil elastase, allowing unchecked degradation of alveolar elastin. The resulting loss of septal architecture produces permanent airspace enlargement and diminishes elastic recoil, hallmarks of emphysematous disease (Kirkham & Barnes, 2013). In parallel, airway-centered remodeling drives the bronchitic phenotype. Persistent activation of epidermal growth factor receptor and inflammatory transcriptional programs promotes goblet cell expansion and excessive mucus production. When combined with oxidative impairment of ciliary motility, this remodeling traps secretions within the airway lumen, producing chronic cough, sputum retention, and recurrent infection (Rahman, 2005). Within this chronically inflamed and genetically unstable environment, malignant transformation becomes increasingly probable. Accumulated DNA lesions and epigenetic dysregulation converge with aberrant antioxidant signaling to facilitate progression from epithelial metaplasia to dysplasia and, ultimately, invasive lung carcinoma (Sporn & Liby, 2012).
10.2 Cardiovascular Injury Through Endothelial Redox Failure
The pathological reach of cigarette smoke extends far beyond the lung. Cardiovascular disease represents one of the most lethal systemic consequences of chronic exposure. Central to this association is endothelial dysfunction, a condition driven by redox imbalance at the vascular interface. Physiological regulation of vascular tone depends on nitric oxide signaling. In smoke-exposed individuals, superoxide derived from circulating oxidants and activated immune cells reacts rapidly with nitric oxide, producing peroxynitrite. This reaction simultaneously depletes a critical vasodilator and generates a highly cytotoxic species that injures endothelial cells. The outcome is impaired vasodilation, increased vascular stiffness, and a pro-inflammatory endothelial phenotype (He & Zuo, 2020). This injury initiates the cascade of atherogenesis. Oxidative modification of low-density lipoproteins promotes their uptake by macrophages, forming foam cells and accelerating plaque development. Over time, this process elevates the risk of myocardial infarction and stroke, establishing cardiovascular disease as a dominant contributor to smoking-related mortality (Dröge, 2002; He & Zuo, 2020).
10.3 Neurobiological Consequences and the Brain–Lung Axis
Increasing evidence supports the existence of a bidirectional neuro-pulmonary axis linking chronic lung inflammation to neurodegenerative disease. Circulating cytokines and reactive species generated in the lung compromise blood–brain barrier integrity, permitting neuroinflammatory signaling within the central nervous system. This process has been implicated in the pathophysiology of disorders such as Alzheimer’s and Parkinson’s disease (Dröge, 2002; Yuan & Yankner, 2000). This biological reality stands in stark contrast to subjective experience. Although smokers frequently report stress relief following nicotine intake, this perception reflects a reversal of withdrawal-induced dysphoria rather than true anxiolysis. Chronic nicotine dependence elevates baseline stress markers, creating a paradox in which the nervous system is simultaneously exposed to oxidative injury and neurochemical destabilization driven by addiction cycles (Parrott, 1999).
10.4 Hepatic, Renal, and Gastrointestinal Involvement
Systemic distribution of smoke-derived toxicants imposes a substantial burden on detoxification organs. The liver, as the primary site of xenobiotic metabolism, experiences oxidative stress through depletion of glutathione reserves and sustained inflammatory signaling. This redox imbalance sensitizes hepatic tissue to secondary insults, promoting inflammatory liver injury (Jaeschke, 2011). The gastrointestinal system emerges as an additional axis of pathology. Inhaled particulates swallowed during smoking, combined with systemic inflammation, disrupt the intestinal microbiome. This dysbiosis weakens epithelial tight junctions, increasing intestinal permeability and permitting microbial metabolites to enter the circulation. The resulting feedback loop amplifies pulmonary inflammation, reinforcing the concept that chronic lung disease reflects a failure of coordinated mucosal immunity across organ systems (Budden et al., 2017; Lynch & Pedersen, 2016).
10.5 Reproductive Toxicity and Developmental Programming
Cigarette smoke constituents extend their influence to reproductive biology. Many components act as endocrine-disrupting chemicals, interfering with hormonal signaling through receptor mimicry or antagonism (Colborn et al., 1993). Exposure during critical developmental windows produces lasting effects, including fetal growth restriction and altered metabolic programming. At the cellular level, oxidative injury to germ cells compromises genomic integrity and reproductive fidelity. Such damage carries intergenerational implications, potentially influencing molecular asymmetry and developmental trajectories fundamental to organismal organization (Blackmond, 2010).
10.6 Endocrine and Metabolic Reprogramming
Chronic inflammation and sustained activation of antioxidant response networks exert profound effects on systemic metabolism. Redox-responsive transcriptional systems interface directly with intermediary metabolic pathways, shifting cellular priorities from energy storage toward continuous defense. This reprogramming disrupts insulin signaling and glucose homeostasis, contributing to the development of type 2 diabetes mellitus (Hayes & Dinkova-Kostova, 2014). Taken together, these changes illustrate the ultimate consequence of prolonged cigarette smoke exposure: a coordinated failure across multiple physiological systems. Respiratory mechanics, vascular integrity, neural regulation, metabolic control, and reproductive health all deteriorate under persistent oxidative and inflammatory pressure, underscoring cigarette smoke as a driver of whole-body pathophysiological collapse rather than isolated organ damage (Agliarulo et al., 2021; Dröge, 2002).
11. Temporal and Systems Chronology of Smoke-Induced Pathogenesis
11.1 Ultrafast Chemical Initiation: Events Below Biological Time
Any realistic model of cigarette smoke induced disease must begin outside the timescale typically associated with biology. The earliest pathogenic events unfold at the level of physical chemistry, long before cellular perception or signaling is possible. The initiating reactions are governed by quantum-mechanical electron transfer, occurring within femtoseconds to picoseconds (10?¹?–10?¹² s). At this scale, semi-stable radicals embedded in the particulate phase engage directly with the solvent environment of the airway lining fluid, following energy landscapes described by Marcus theory (Marcus, 1993). Once solvated, these radicals participate in reaction cascades limited not by enzymatic regulation but by molecular diffusion. A defining example is the near-diffusion-controlled interaction between nitric oxide and superoxide, yielding peroxynitrite with a rate constant on the order of 10? M?¹·s?¹ (Pryor & Stone, 1993). On any biological timescale, this reaction is effectively instantaneous. The implication is profound: formation of highly cytotoxic species precedes the activation of antioxidant enzymes or stress-response signaling. The earliest “hit” therefore occurs within microseconds, irreversibly reshaping the local redox landscape before the cell can register the presence of danger.
11.2 Early Cellular Response: The Critical Window of Signaling
Following the initial chemical insult, biological processes begin to unfold on a timescale accessible to cellular machinery. The period spanning minutes to hours constitutes a decisive signaling interval in which cellular fate is largely determined. Oxidative modification of redox-sensitive cysteine residues serves as the first intracellular alarm, rapidly altering protein activity and initiating redox-based signaling cascades (Sies & Jones, 2020). Within this window, depletion of intracellular glutathione and direct mitochondrial membrane injury promote release of ATP into the extracellular space. Extracellular ATP functions as a damage-associated signal, engaging purinergic receptors and driving calcium influx alongside potassium efflux. These ionic disturbances, coupled with sustained reactive species generation, trigger phosphorylation and degradation of inhibitory regulators of inflammatory transcription factors. By approximately one hour post-exposure, nuclear accumulation of NF-κB is well established, while parallel oxidative stabilization of Nrf2 initiates transcription of cytoprotective genes (Rahman, 2005). This interval represents a cellular decision point—a kinetic competition between inflammatory escalation and adaptive defense. Under physiological stress, these pathways remain balanced. Under cigarette smoke exposure, the magnitude and persistence of oxidant input frequently overwhelm protective induction, biasing the system toward inflammation and setting the trajectory for chronic pathology.
11.3 Intermediate Adaptation and the Point of Irreversibility
With continued exposure over days to weeks, the system transitions from acute response to structural reorganization. Persistent oxidative injury to proteins and organelles activates autophagy, the primary mechanism for intracellular quality control (Mizushima et al., 2008). Initially compensatory, this process becomes progressively inefficient as the rate of damage exceeds clearance capacity. The result is accumulation of misfolded proteins and dysfunctional mitochondria that continue to generate reactive species. During this period, cellular senescence emerges as a dominant outcome. Cells experiencing extensive DNA damage and telomere attrition withdraw permanently from the cell cycle. Although senescence initially functions as a tumor-suppressive mechanism, senescent cells acquire a secretory phenotype characterized by sustained release of inflammatory cytokines, proteases, and matrix-degrading enzymes (Campisi & d’Adda di Fagagna, 2007). This marks a critical biological inflection point. A mechanism designed to prevent malignant transformation becomes a source of chronic tissue injury. In the lung, this transition underlies progressive parenchymal destruction and airway remodeling, embedding inflammation and structural loss into the tissue architecture.
11.4 Long-Term Amplification and Multisystem Failure
The final phase of the pathogenic timeline unfolds over months to years and parallels processes associated with aging. Accumulation of senescent cells, exhaustion of antioxidant transcriptional programs, and stable epigenetic activation of inflammatory genes collectively drive irreversible disease states. In the respiratory system, slow but continuous degradation of alveolar structure culminates in chronic obstructive pulmonary disease, reflecting an accelerated manifestation of oxidative aging predicted decades earlier by free radical theories of degeneration (Harman, 1956). In the vascular system, sustained oxidative modification of lipoproteins and endothelial dysfunction promotes atherogenesis, eventually manifesting as ischemic cardiovascular events. Most critically, persistent genomic injury combined with selective survival of cells harboring dysregulated antioxidant signaling enables the multistep evolution of cancer. Over time, clones with enhanced resistance to oxidative stress and apoptosis are favored, completing the transition from chronic injury to malignancy (Hanahan & Weinberg, 2011). Thus, the entire disease continuum from quantum-scale electron transfer to organism-level failure can be traced along a single temporal axis. What begins as a femtosecond chemical event is progressively amplified through biological time, ultimately presenting as the complex, lethal syndromes associated with long-term cigarette smoke exposure.
12. Computational Integration of Redox-Bioenergetic Pathology
12.1 Multi-Scale Modeling of Redox and Bioenergetic Collapse
The pathological cascade initiated by cigarette smoke unfolds across temporal and spatial scales that exceed intuitive human synthesis. Events range from femtosecond electron transfer reactions to disease phenotypes emerging over decades. Bridging these scales necessitates a computational framework capable of integrating physical chemistry, cellular kinetics, and systems physiology. This role is fulfilled by modern in silico pharmacology, which has matured from isolated molecular simulations into a discipline encompassing systems-level toxicology (Ekins et al., 2007). The central aim is the construction of a computational “digital twin” of the redox–bioenergetic axis. This requires hierarchical integration of multiple modeling layers. At the molecular level, quantum mechanical calculations such as density functional theory define radical energetics, electron density redistribution, and bond dissociation parameters. These outputs inform molecular dynamics simulations that resolve conformational transitions in redox-sensitive proteins, including stress sensors and transcriptional regulators. Molecular events are subsequently embedded within kinetic frameworks based on ordinary differential equations, allowing simulation of intracellular reaction fluxes under defined oxidative loads. This multi-scale coupling enables forward prediction of how discrete chemical insults propagate through metabolic and signaling networks to produce emergent cellular behaviors.
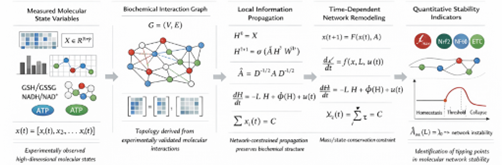
Figure 12.1 Graph-theoretic and dynamical systems framework formalizing redox–bioenergetic network propagation, exposure-driven state evolution, and stability thresholds governing transition from molecular homeostasis to pathological collapse.
12.2 Mixture-of-Experts Architectures for Organ-Specific Responses
Biological responses to cigarette smoke are intrinsically heterogeneous. Identical oxidative inputs elicit divergent outcomes across epithelial, immune, and vascular compartments. This heterogeneity renders single, monolithic neural architectures inadequate. Instead, we adopt a mixture-of-experts framework, in which the overall system is decomposed into specialized computational modules. Each expert network is trained to represent a distinct biological domain, such as airway epithelial integrity, macrophage immune-metabolic activation, or endothelial redox regulation. A supervisory gating network dynamically integrates these expert outputs, assigning context-dependent weights based on exposure patterns, duration, and tissue state. This architecture permits simulation of inter-organ communication and systemic spillover while maintaining interpretability and computational efficiency (Shazeer et al., 2017).
12.3 Graph Neural Networks for Redox and Signaling Topology
Cellular systems operate as interaction networks rather than linear pathways. To capture this relational structure, we represent molecular and protein interactions as graphs, analyzed using graph neural networks. In this representation, reactive species, metabolites, transcription factors, and receptors constitute nodes, while biochemical reactions, binding events, and regulatory interactions define edges. Graph convolutional networks are employed to learn structural features embedded within this topology, enabling identification of densely connected modules and signal propagation routes (Kipf & Welling, 2017). To resolve context-dependent importance, graph attention mechanisms assign adaptive weights to interactions, allowing the model to prioritize specific edges under defined stress conditions (Veli?kovi? et al., 2018). This approach enables computational identification of regulatory hubs such as redox sensors, purinergic receptors, or inflammasome components that disproportionately influence system stability. It provides algorithmic support for the principles of network pharmacology, where therapeutic leverage emerges from nodal control rather than single-target inhibition (Hopkins, 2008; Wu & Li, 2019).
12.4 Temporal Learning and the Encoding of Biological Memory
Static network representations cannot capture the temporal dimension that defines disease progression. To model dynamic behavior, we incorporate sequence-learning architectures capable of encoding history-dependent responses. Recurrent neural networks, particularly long short-term memory units, are used to model how prior exposures modify baseline cellular responsiveness. These models simulate biological memory, including cumulative oxidative burden and epigenetic conditioning (Hochreiter & Schmidhuber, 1997). For longer temporal dependencies spanning early molecular damage to late phenotypic outcomes we employ transformer architectures. The self-attention mechanism inherent to transformers enables direct coupling of temporally distant events, allowing the model to associate early redox disruptions with downstream outcomes such as senescence, metabolic reprogramming, or malignant transformation (Vaswani et al., 2017). This effectively transforms disease modeling into a sequence-reading problem, where the system interprets the life history of the cell.
12.5 Physics-Informed Constraints and Mechanistic Fidelity
Purely data-driven models risk generating predictions that violate fundamental physical or chemical laws. To prevent such artifacts, we integrate physics-informed neural networks into the modeling pipeline. In these systems, governing equations derived from thermodynamics, reaction kinetics, and mass conservation are embedded directly into the training objective. By penalizing solutions that violate established principles such as conservation of matter, energetic feasibility, or reaction stoichiometry these constraints enforce mechanistic plausibility. This hybrid approach unites empirical learning with first-principles modeling, ensuring that predictions remain biologically and chemically admissible (Raissi et al., 2019).
CONCLUSION
Cigarette smoke-driven disease is not the product of random molecular injury or the accumulation of independent risk factors. It is the predictable consequence of sustained destabilization within tightly regulated biological networks. The initiating events are chemical and physical, unfolding at timescales far faster than cellular perception. These early radical reactions irreversibly distort the redox environment, setting in motion a cascade in which bioenergetic integrity erodes, antioxidant governance fails, and immune signaling becomes chronically misaligned. With continued exposure, these coupled disruptions are consolidated through epigenetic fixation, cellular senescence, and clonal selection, ultimately manifesting as chronic organ dysfunction and malignant transformation. The historical failure of antioxidant-based interventions reflects a fundamental misinterpretation of redox biology. Reactive species are not expendable by-products but essential regulatory signals. Non-specific suppression therefore dismantles physiological control rather than restoring balance. Effective intervention must instead focus on preserving and reconstituting system resilience by reinforcing endogenous defense capacity, constraining pathological sources of oxidants, and interrupting the feedback loops that convert transient injury into persistent inflammation.
Clinical disease, in this framework, represents the late expression of earlier, quantifiable network failure rather than an abrupt pathological event. Therapeutic success is therefore governed more by timing than by intensity, requiring action during adaptive phases before regulatory systems become irreversibly reprogrammed. Because these failures arise from interacting pathways rather than single defects, meaningful prevention and treatment must operate at the network level, aligning intervention strategies with the architecture of biological regulation itself. In essence, cigarette smoke acts as a systems-level perturbation, and disease constitutes the long-term biological memory of unresolved stress. The path forward lies in predictive modeling, network-based pharmacology, and early preventive strategies that prioritize the maintenance of regulatory integrity over attempts to repair collapse once it has already occurred.
REFERENCES

Tushar Sharma, Nutan Sahu, Dr. Gyanesh Kumar Sahu, Redox–Bioenergetic Network Collapse in Cigarette Smoke Exposure: Molecular Mechanisms Linking Radical Chemistry to Genomic Instability, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 191-216. https://doi.org/10.5281/zenodo.19383648
 10.5281/zenodo.19383648
10.5281/zenodo.19383648
