College of Pharmaceutical Sciences, Govt. Medical College, Kozhikode
Conventional oral drug delivery systems often face challenges such as premature drug release, inconsistent absorption, and difficulty in targeting multiple regions of the gastrointestinal (GI) tract. To overcome these limitations, the present study developed a Rocket Inspired Multi-site Oral Tablet (RIMOT) designed for the targeted delivery of Loperamide Hydrochloride and Simethicone. The design concept was adapted from the stage-separation mechanism of multi-stage rockets, where each part detaches sequentially. Similarly, RIMOT was engineered to enable controlled layer separation, ensuring timely and site-specific drug release. The system consists of three functional layers. The floating layer, formulated using hydroxypropyl methylcellulose (HPMC) and sodium bicarbonate, is intended to remain buoyant in gastric fluid and release drug locally. The intermediate microsphere-based layer contains super disintegrants that facilitate rapid disintegration in gastric conditions, enabling efficient separation of the adjacent layers and ensuring controlled intestinal drug release with high entrapment efficiency. The colon-targeted layer, prepared using guar gum, resists drug release in the stomach and intestine but enables selective release in colonic media. The optimized formulations demonstrated desirable physicomechanical properties, uniform drug content, and satisfactory in-vitro release behaviour. The floating layer showed a short lag time of 32 seconds and buoyancy exceeding 7 hours, while the microsphere layer ensured controlled intestinal delivery, and the colon layer released drug specifically under colonic conditions. Overall, RIMOT was successfully developed as an innovative multisite delivery system. Its rocket-inspired separation mechanism enhances therapeutic outcomes for conditions such as traveller’s diarrhoea and flatulence and offers a versatile platform adaptable to other drugs requiring targeted GI delivery.
Delivering drugs to different sites within the GI tract is inherently complex and calls for innovative approaches (1). The distinctive features of the gastrointestinal tract need to be considered when developing drugs for oral delivery. Variations in boundary conditions across different GI compartments including differences in surface area, pH, cellular composition, transport mechanisms, permeability, and microbial populations create both obstacles and possibilities for site-specific drug delivery (2). Despite advancements in multi-site drug delivery systems, existing technologies often struggle with achieving precise, sequential drug release at targeted regions of the gastrointestinal (GI) tract. Most conventional approaches, such as multiparticulates or pulsatile systems, face limitations in terms of dose accuracy, inconsistent release patterns, and sensitivity to GI conditions like pH, motility, and fluid composition (3) .These drawbacks can lead to suboptimal therapeutic effects, especially for drugs that require both localized action and controlled release at multiple sites (4). To overcome the inherent limitations of conventional and current multi-site oral drug delivery systems, the Rocket-Inspired Multi-site Oral Tablet (RIMOT) was conceptualized (5). This novel system draws its core design from the multi-stage rocket separation mechanism used in space exploration. In a multi-stage rocket, different sections or “stages” are strategically separated to perform specific tasks, each stage detaches allowing the next to continue its journey efficiently without added burden (6). Similarly, RIMOT is structured as a three-layered oral tablet, each layer intended for drug release at a distinct site within the gastrointestinal (GI) tract. RIMOT consist of
The middle layer is positioned between the upper floating layer and the lower colon-targeted layer. Middle layer is formulated with super disintegrants and drug-loaded microspheres, rapidly disintegrates upon contact with gastrointestinal fluids. This disintegration facilitates the physical separation of the upper and lower layers, enabling each to follow its intended path within the gastrointestinal tract. The microspheres embedded within the middle layer are for targeted drug release in the small intestine, exploiting favorable pH and enzymatic conditions for optimal absorption. Once the middle layer disintegrates, the floating layer stays buoyant in the stomach to provide sustained or localized release, while the bottom pH-sensitive layer remains intact through the small intestine and releases the drug only in the colon. To effectively demonstrate the RIMOT system, loperamide HCl and simethicone were selected as model drugs. In traveler’s diarrhea accompanied by bloating, this combination is commonly used—simethicone provides gastric retention through the floating layer, while loperamide, which acts on intestinal smooth muscles, is incorporated into microspheres and the colon-targeted layer for localized intestinal action. Thus, RIMOT provides site-specific delivery of therapeutic agents, mimicking the strategic release seen in rocket separation systems—ensuring improved efficacy, better localization, and minimal systemic side effects
MATERIALS AND METHODS
MATERIALS
Loperamide hydrochloride was obtained from Yarrow Chem Products, Mumbai, and simethicone was sourced from RioCare India Private Limited, Mumbai, while all other excipients used in the formulation were of pharmaceutical grade and procured from reputable suppliers.
METHODS
Analysis of drug
Physical appearance of drug
The drug powder was observed visually for physical appearance and homogeneity (7).
Identification of the drug by FTIR spectroscopic analysis
FTIR spectroscopy was utilized to identify the drug by analysing the position and intensity of characteristic absorption band. Spectral data were recorded within the range of 4000–700 cm?¹ using an FTIR instrument equipped with an ATR accessory
Determination of λmax and preparation of calibration curve of Loperamide HCl
10 mg of Loperamide HCl was solubilized in a minimal volume of 0.1 N hydrochloric acid using 0.5 ml of Tween 80, and the solution was diluted to 100 ml with 0.1 N hydrochloric acid in a volumetric flask. Aliquots of 0.2, 0.4, 0.6, 0.8, and 1 ml were accurately transferred into separate volumetric flasks and diluted to 10 ml with 0.1 N hydrochloric acid. Absorbance readings were recorded at 220 nm using a UV spectrophotometer, and a calibration curve was constructed by plotting concentration against absorbance.
Formulation Of Colon Targeted Tablet
The colon-targeted layer (third layer) was prepared by direct compression as per Table 1. Loperamide HCl, guar gum, lactose, and microcrystalline cellulose were sieved and blended, followed by addition of magnesium stearate and talc. The final blend was compressed using a 6.35 mm punch. (8,9)
Table 1: Formulation of colon targeted tablet layer
|
Ingredients |
F1 (mg) |
F2 (mg) |
F3 (mg) |
|
Loperamide HCl |
2 |
2 |
2 |
|
Guar gum |
20 |
30 |
40 |
|
Microcrystalline cellulose |
50 |
40 |
30 |
|
Lactose |
20 |
20 |
20 |
|
Magnesium stearate |
3 |
3 |
3 |
|
Talc |
5 |
5 |
5 |
|
Total weight |
100 mg |
100 mg |
100 mg |
Precompression Parameters
Precompression studies were carried out to assess the flow properties of the powder blend. The evaluated parameters included angle of repose, bulk density, tapped density, Carr’s index, and Hausner’s ratio, which help determine powder flowability and suitability for compression (10)
Estimation of Tablet Physical Parameters
The formulated tablets were examined for their physical qualities, including thickness, hardness, friability, and drug content. Thickness, hardness, and friability were evaluated to confirm uniform tablet dimensions and adequate mechanical strength. Thickness was measured using a screw gauge, hardness was checked with a tablet hardness tester, and friability was determined using a Roche friabilator. Drug content uniformity was assessed by selecting ten tablets at random, powdering them, dissolving an accurately weighed portion equivalent to the required drug amount in 0.1N HCl, and measuring the absorbance at 220 nm to verify uniform distribution of the drug (11).
In vitro Drug Release of colon targeted tablet
In vitro release of Loperamide HCl from the colon-targeted tablets was studied using a USP Type II apparatus in 900 ml of dissolution medium at 37 ± 0.5 °C and 50 rpm. Samples were collected at predetermined intervals as the medium was sequentially changed from 0.1 N HCl to phosphate buffer pH 6.8 and then to phosphate buffer pH 7.4. All samples were analysed spectrophotometrically at 220 nm(11) .
Formulation Loperamide HCl-Loaded Microspheres
(Ionic Gelation Method)
Loperamide-loaded microspheres were prepared by dispersing sodium alginate (1%, 1.5%, and 2% w/v) in distilled water and stirring for 30 minutes. From each solution, 20 mL was taken and mixed with loperamide hydrochloride. The mixture was added dropwise into 2% w/v calcium chloride solution to form microspheres by ionic crosslinking. The beads were kept in the calcium chloride solution for 1 hour, then filtered and dried at 50 °C for 1 hour(12).
Evaluation of Loperamide-Loaded Microspheres
Scanning Electron Microscopy (SEM)
The shape and surface characteristics of the microspheres were examined using SEM. Samples were placed on carbon tape, vacuum-dried, gold-coated, and imaged to observe surface morphology(13).
Percentage Yield
The yield was calculated by weighing the dried microspheres from each batch and comparing it with the total weight of drug and excipients used (14).
Percentage Drug Content
A quantity of microspheres equivalent to 2 mg of drug was crushed, dissolved in 0.1N HCl, sonicated for 30 minutes, filtered, and analysed at 220 nm to determine drug content(15).
Percentage Entrapment Efficiency
Entrapment efficiency was determined by comparing the actual drug content in the microspheres with the theoretical amount(15).
In Vitro Drug Release
Drug release from the beads was tested using a USP Type II apparatus. Beads containing 2 mg of Loperamide HCl were placed in a dialysis membrane and immersed in 900 mL of 0.1 N HCl at 37 ± 0.5 °C and 50 rpm. Samples were taken after 2 hours and analysed at 220 nm. The medium was then changed to pH 6.8 phosphate buffer for 3 hours. Samples were again analysed at 220 nm(16).
Formulation and Evaluation of Floating Tablet Layer
Floating tablets of Simethicone (adsorbed on magnesium carbonate, 1:1.5) were prepared by direct compression using HPMC, sodium bicarbonate, citric acid, and MCC, followed by lubrication with magnesium stearate and talc. Three formulations were developed by varying HPMC levels (60, 80, and 100 mg) while keeping other ingredients constant. The powder blend was assessed for pre-compression properties, and tablets were compressed and evaluated for standard post-compression parameters(17).
Table 2: Formulation of Floating Tablet Of Simethicone
|
Ingredients |
Formulation (F1) (mg) |
Formulation (F2) (mg) |
Formulation (F3) (mg) |
|
Simethicone: magnesium carbonate |
80:120 |
80:120 |
80:120 |
|
HPMC K4 |
80 |
100 |
120 |
|
Sodium bicarbonate |
120 |
100 |
80 |
|
Citric acid |
10 |
10 |
10 |
|
Microcrystalline cellulose |
10 |
10 |
10 |
|
Magnesium stearate |
3 |
3 |
3 |
|
Talc |
5 |
5 |
5 |
|
Total weight |
410 mg |
410 mg |
|
Pre-compression Studies
The powder blends prepared for the floating layer were assessed for bulk density, tapped density, Carr’s index, Hausner’s ratio, and angle of repose.
Post-compression Parameters
The compressed floating tablets were examined for hardness, thickness, and friability using the methodology previously described
In Vitro Buoyancy Study
Buoyancy performance was assessed by determining the floating lag time (FLT) and total floating time (TFT). Each tablet was placed in 200 mL of 0.1 N HCl, and the time taken to emerge on the surface was recorded as FLT, while the duration it remained afloat was noted as TFT (18).
Fabrication Of RIMOT
RIMOT tablets were prepared by sequential layer-by-layer compression. First, the optimized colon-targeted layer was compressed using a 6.35 mm flat-faced punch. The microsphere blend (microspheres, sodium croscarmellose, and MCC) was then carefully added over this layer and compressed using an 11.11 mm punch. Finally, the floating layer was placed on top of the microsphere layer and compressed with the same 11.11 mm punch to obtain the final trilayer RIMOT tablet.
Evaluation of RIMOT
The RIMOT tablets were evaluated for post-compression parameters such as thickness, hardness, and friability.
Drug Content Uniformity
Five tablets were powdered, and an amount equivalent to 4 mg of drug was dissolved in 0.1 N HCl. The mixture was sonicated for one hour, filtered, and the filtrate was analyzed at 220 nm using a UV–visible spectrophotometer (19).
In Vitro Drug Release
Drug release was studied using a USP Type II dissolution apparatus. Tablets were placed in 900 mL of 0.1 N HCl at 37 ± 0.5 °C and 50 rpm. Samples were taken after 2 hours. The medium was then replaced with phosphate buffer pH 6.8 for 3 hours, followed by phosphate buffer pH 7.4 for 2 hours. Absorbance was measured at 220 nm to obtain the release profile (20).
In Vitro Buoyancy Test
Floating lag time and total floating time were measured by placing each tablet in 200 mL of 0.1 N HCl and observing its floating behavior (18).
RESULTS AND DISCUSSION
Analysis of drug
Physical appearance of drug
It was discovered that loperamide hydrochloride is a white powder that matches the claimed medication description of drug.
Drug identification using infrared spectroscopy
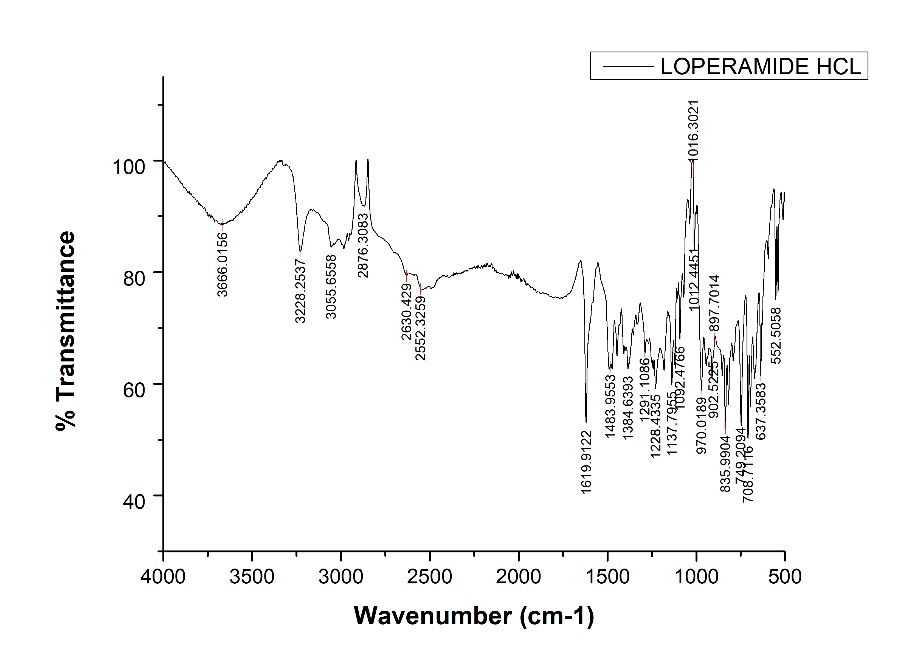
Figure 1: FTIR Spectrum of Loperamide HCl
Table 3: Spectroscopic peaks of loperamide HCl
|
Wave Number cm−1 |
Functional Group |
|
1037 |
C-Cl stretch |
|
1384 |
CH3 group |
|
1483 |
-CO stretch |
|
1619 |
C=O stretch |
|
2876 |
CH stretch |
|
3228 |
O-H stretch |
FT-IR spectra of the pure drug sample contained all of the distinctive vibrations that corresponded to the functional groups of Loperamide HCl (Figure 5.2, Table 5.2). The spectrum thus met reference standard.
Determination of λmax of Loperamide hydrochloride
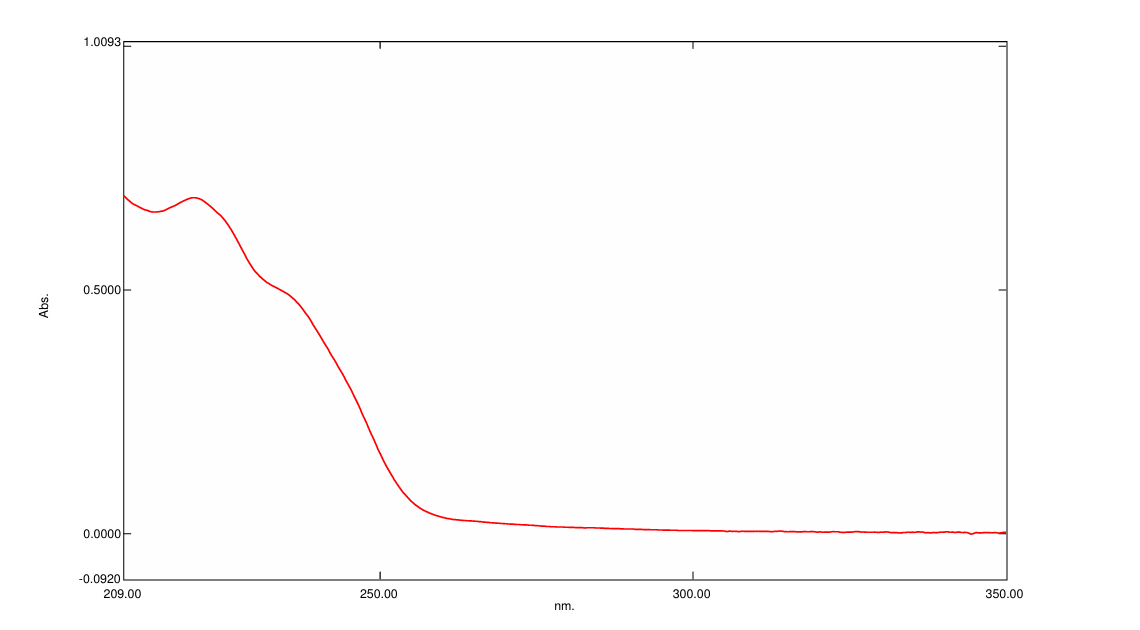
Figure 2: λmax of Loperamide hydrochloride
Calibration plot of Loperamide Hydrochloride in 0.1 N HCl
Table 4: Concentration and absorbance obtained for calibration curve of Loperamide HCl
|
Concentration |
Absorbance |
|
2 |
0.1241 |
|
4 |
0.2163 |
|
6 |
0.3701 |
|
8 |
0.5147 |
|
10 |
0.6855 |
|
12 |
0.859 |
|
14 |
0.938 |
Calibration curve of Loperamide HCl in 0.1N HCl pH 1.2 was plotted using different concentration of drug (2, 4, 6,8,10 µg/ml) and by measuring absorbance at 220 nm.
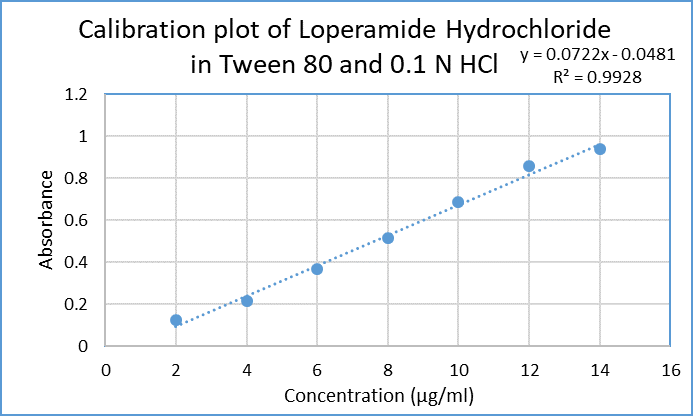
Figure 3: Calibration curve of Loperamide HCl in 0.1 N HCl
Evaluation of colon targeted tablet layer of loperamide hydrochloride
Precompression study
Table 5: Precompression study of colon targeted tablet
|
Parameters |
F1 |
F2 |
F3 |
|
Bulk density (g/ml) |
0.418±0.032 |
0.424±0.021 |
0.434±0.012 |
|
Tapped density (g/ml) |
0.5±0.041 |
0.52±0.009 |
0.55±0.036 |
|
Hausner’s ratio |
1.19±0.028 |
1.22±0.010 |
1.26±0.016 |
|
Compressibility index% |
16.8±0.031 |
18.4±0.013 |
20±0.019 |
|
Angle of repose (θ) |
32.7±1.07 |
34.5±0.97 |
37±0.081 |
The powder blend exhibited acceptable bulk and tapped density values. The compressibility index and Hausner’s ratio indicated good compressibility and flow behavior. The angle of repose further confirmed that the blend possessed passable flow properties.
Thickness, Hardness, and Friability
All three formulations (F1, F2, and F3) showed uniform thickness, adequate hardness, and friability values within acceptable limits. These results indicate good consistency in tablet compression and satisfactory mechanical integrity. The detailed numerical values are provided in Table 4
Table 6: Post compression parameters of colon targeted tablet
|
Parameters |
F1 |
F2 |
F3 |
|
Thickness (mm) (±SD) |
3.1±0.01 |
3.1±0.93 |
3.2±0.006 |
|
Hardness (kg/cm2) (±SD) |
5.65±0.08 |
5.32±0.05 |
5.3±0.12 |
|
Friability (%) (±SD) |
0.39±0.86 |
0.46±0.62 |
0.68±1.10 |
|
Durg content uniformity (%) (±SD) |
87.60±1.91 |
90.1±0.09 |
89.5±1.26 |
Drug content uniformity
The drug content of the three tablet formulations was found to be 87.60±1.91, 90.1±0.09 and 89.5±1.26 for F1, F2 and F3 respectively (Table 4). All values were within the acceptable pharmacopeial limits, indicating that the formulations contained a reasonably uniform distribution of the active drug.
In vitro drug release
F3, with the lowest %CDR of 49.9±0.986 % at 7 hours, was selected for colon targeting due to its sustained release profile and minimal premature drug release (Table 5)
Table 7: In vitro dissolution data of loperamide HCl colon targeted tablet
|
Time (h) |
% CDR of F1 |
% CDR OF F2 |
% CDR OF F3 |
|
2 |
20±0.156 |
15.9±0.135 |
13.2±±0.235 |
|
5 |
44.5±1.02 |
36.9±0.962 |
34.1±0.411 |
|
7 |
64.9±1.21 |
55.6±1.08 |
49.9±0.986 |
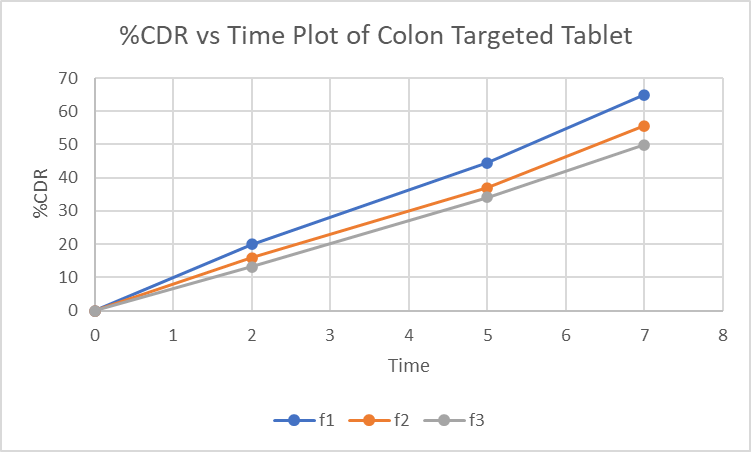
Figure 4: %CDR vs Time graph of Loperamide HCl in 0.1 N HCl, pH 6.8 phosphate buffer and pH 7.4 phosphate buffer
In vitro drug release kinetics study of selected formulation F3 of Colon targeted tablet
The in vitro drug permeation data were analysed for goodness of fit using linear regression, applying various kinetic models such as zero-order, first-order, Higuchi, and Korsmeyer–Peppas (Figures 2–5) to determine the drug release mechanism. The outcomes of the regression analysis, including the corresponding correlation coefficients, are summarized in table 6.
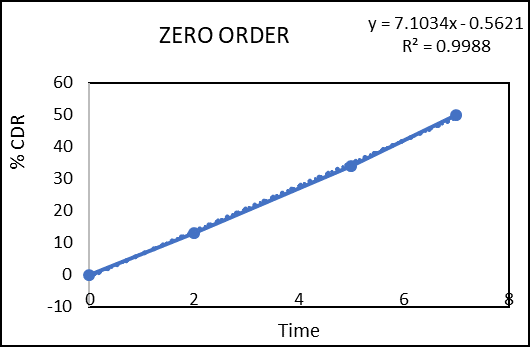
Figure 5: Zero order kinetic model of colon targeted tablet formulation of F3
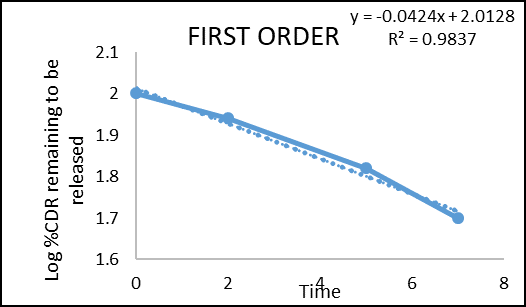
Figure 6: First order kinetic model of colon targeted tablet formulation of F3
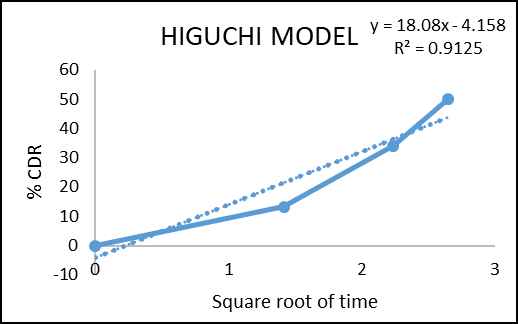
Figure 7: Higuchi model of colon targeted tablet formulation of F3
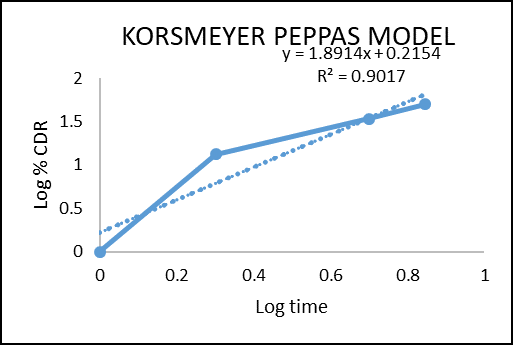
Figure 8: Korsmeyer Peppas model colon targeted tablet formulation of F3
Table 8: Regression coefficient of various kinetic models of selected f3 formulation of colon targeted tablet
|
Zero Order |
First Order |
Higuchi Model |
Korsmeyer Peppase Model |
|
|
R2 |
R2 |
R2 |
R2 |
N |
|
0.9988 |
0.9837 |
0.9125 |
0.9017 |
1.891 |
Regression coefficient (R2) obtained for zero order kinetics and first order kinetics of F3 were 0.9988 and 0.9837 respectively. The result indicates that the release governed by zero order kinetics. The obtained n value from the Korsmeyer–Peppas model was 1.891 (n > 1), which indicates Super Case II transport reflecting that swelling and erosion are the major governing factors.
Preparation and evaluation of microsphere of Loperamide HCl
Three formulations were prepared using different sodium alginate concentrations (1%, 1.5%, and 2% w/v) with a fixed 2% w/v calcium chloride. The 1% alginate failed to form proper microspheres due to inadequate crosslinking, while the 1.5% and 2% formulations produced well-formed microspheres through effective ionic gelation.
Surface morphology and Particle shape


Figure 9: SEM image of microspheres a) F2, b) F3
The concentration of sodium alginate influenced microsphere morphology. The lower alginate level in F2 produced weak, irregular, and porous microspheres, while the higher concentration in F3 formed stronger, smoother, and more spherical microspheres. Thus, increasing alginate concentration improves shape, reduces porosity, and supports better drug entrapment and controlled release.
Percentage Yield
The percentage yield increased with higher alginate concentration, with F2 showing 84.6±1.06% and F3 achieving 96.8±0.07%, due to improved crosslinking, bead stability, and reduced material loss.
Table 9: Evaluation of Loperamide HCl Microspheres
|
Formulation |
% Yield |
% Drug content |
Entrapment efficiency |
|
F2 |
84.6±1.06 |
85.7±1.27 % |
80.90±0.98 % |
|
F3 |
96.8±0.07 |
93.1±0.09% |
89.8±0.13 % |
Drug Content
F3 showed higher drug content (93.1±0.09%) compared to F2 (85.7±1.27%), as presented in Table 7
Entrapment Efficiency
F3 also exhibited higher entrapment efficiency (89.8±0.13%) than F2 (80.9±0.98%) due to the stronger gel network formed by its higher alginate concentration (Table 7).
In vitro Drug release
The drug release from microsphere F2 was 46.1±0.27% at 2?h and 90±1.38% at 5?h, while F3 showed 38±0.94% at 2?h and 89.2±1.27% at 5?h (table 8).F3 exhibited slower initial release and more controlled drug release compared to F2, making it the finalized formulation to prevent premature drug release in the stomach.
Table 10: In vitro drug release of microsphere
|
Time (h) |
% CDR OF F2 |
% CDR OF F3 |
|
2 |
46.1±0.27 |
38±0.94 |
|
5 |
90±1.38 |
89.2±1.27 |
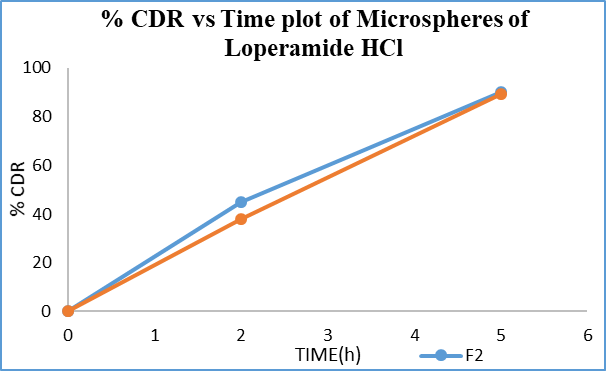
Figure 10: % CDR vs Time graph of loperamide HCl containing microspheres
In vitro release kinetics of selected microsphere formulation F3
To assess goodness of fit, the in vitro drug release data were subjected to linear regression analysis using multiple kinetic models (fig 8-11).
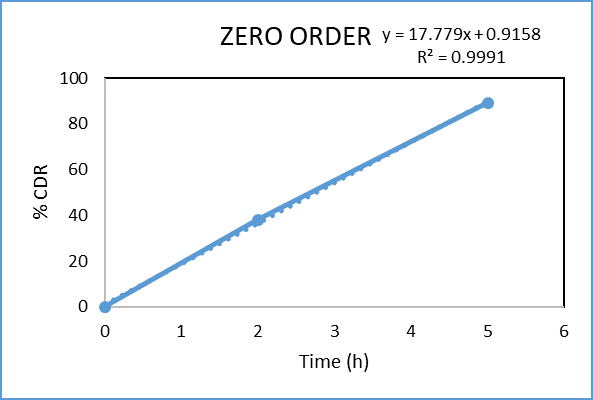
Figure 11: Zero order kinetic model of microspheres of formulation F3
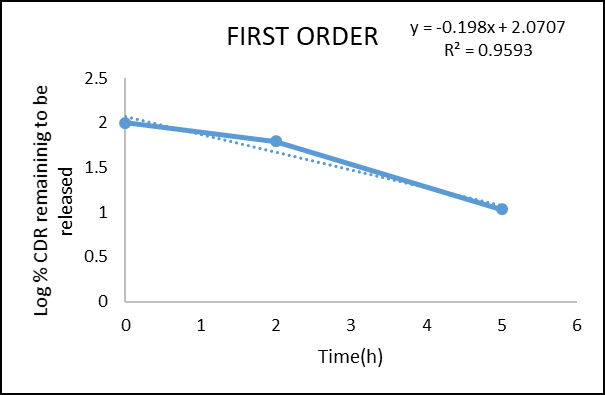
Figure 12: First order kinetic model of microsphere formulation of F3

Figure 13: Higuchi model of microsphere formulation of F3

Figure 14: Korsmeyer Peppas model of microsphere formulation of F3
Table 11: Regression coefficient of various kinetic models of microsphere formulation f3
|
Zero Order |
First Order |
Higuchi Model |
Korsmeyer Peppas Model |
|
|
R2 |
R2 |
R2 |
R2 |
n |
|
0.9991 |
0.9593 |
0.9449 |
0.831 |
2.693 |
Regression coefficient (R²) obtained for zero-order and first-order kinetics of the prepared microsphere tablet were 0.9991 and 0.9593, respectively. The result confirm that the drug release obeys zero-order.The obtained n value from the Korsmeyer Peppas model was 2.693 (n > 1), which shows that the mechanism of drug release follows Super Case II transport, indicating that swelling and erosion are the major governing factors.
Preparation and evaluation floating tablet layer of Simethicone

Figure 15: Floating tablet layer of Simethicone
Precompression study of floating tablet
Table 12: precompression parameters of floating tablet
|
Parameters |
F1 |
F2 |
F3 |
|
Bulk density (g/ml) (± S.D) |
0.35±0.061 |
0.368±0.043 |
0.38±0.05 |
|
Tapped density (g/ml) |
0.411±0.019 |
0.437±0.012 |
0.46±0.037 |
|
Hausner’s ratio (± S.D) |
1.17±0.037 |
1.19±0.041 |
1.22±0.056 |
|
Compressibility index% (± S.D) |
14.8±0.006 |
16.7±0.04 |
17.3±0.05 |
|
Angle of repose (θ) (± S.D) |
36.2±0.02 |
37.5±0.36 |
38.9±0.07 |
The pre-compression flow properties of all three formulations showed a slight decrease in flowability with increasing HPMC concentration, yet remained within acceptable limits for direct compression, as detailed in Table 10.
Post compression parameters of floating tablet
The post-compression results showed consistent thickness, adequate hardness, and decreasing friability with higher HPMC concentration, all remaining within acceptable limits and indicating good mechanical strength, as presented in Table 11.
Table 13: Post compression study of floating tablet
|
Parameters |
F1 |
F2 |
F3 |
|
Thickness (mm) |
4.47±0.06 |
4.51±0.13 |
4.59 ±0.25 |
|
Hardness (kg/cm2) |
5.0 ±1.06 |
5.24±0.96 |
5.36 ±0.64 |
|
Friability (%) |
0.9±0.08 |
0.64±0.12 |
0.45±0.15 |
In vitro buoyancy test of floating tablet
The buoyancy study revealed that the floating lag time (FLT) decreased by increasing polymer concentration. F1 exhibited an FLT of 66±0.008 seconds, F2 showed 43±0.012 seconds, and F3 had the shortest FLT of 29±0.135seconds. The total floating time (TFT) for all the formulations was observed to be greater than 7 hours, indicating satisfactory buoyancy characteristics.
Table 14: In vitro buoyancy test of floating tablet
|
Parameters |
F1 |
F2 |
F3 |
|
Floating lag time(s) |
66±0.008 s |
43±0.012 s |
29±0.135s |
|
Total floating time (h) |
> 7 h |
> 7 h |
>7 h |
Fabrication and Evaluation of Rocket-Inspired Multi-site Oral Tablet
For the fabrication of RIMOT tablets, the colon-targeted tablet was first prepared using the selected F3 formulation and compressed with a 6.35 mm flat-faced punch. This tablet layer was then placed centrally in an 11.11 mm die cavity, over which the optimized microsphere formulation (f3), equivalent to 2 mg of drug was blended with 70 mg of croscarmellose sodium and 20 mg of microcrystalline cellulose and compressed to form the second layer. Finally, the selected floating layer formulation (F3 of simethicone) was placed above the microsphere layer and compressed using the same 11.11 mm punch to obtain the final trilayer RIMOT tablet.


Figure 16: Prepared tablet of RIMOT
Post-compression parameters of RIMOT
The RIMOT tablets were evaluated for post-compression parameters such as thickness, hardness, and friability. The thickness of the tablets was found to be 5.4±0.72mm, indicating uniformity in tablet dimensions. The hardness was observed to be 5.7 ±0.03 kg/cm², which confirms that the tablets possessed adequate mechanical strength to withstand handling without breakage. Friability was determined to be 0.7±0.087%, which is well below the acceptable limit of 1%, signifying good mechanical resistance and suitability of the prepared tablets for further studies
Table 15: Post-compression study of RIMOT
|
Parameters |
Values |
|
Thickness (mm) |
5.4±0.72 |
|
Hardness (kg/cm2) |
5.7±0.03 |
|
Friability (%) |
0.7±0.087 |
Drug content uniformity of RIMOT
The obtained value of drug content uniformity was found to be 92.4±0.142% which falls well within this limit, confirming that the RIMOT tablets contain a uniform distribution of the drug.
In vitro buoyancy test of RIMOT
Upon immersion of the RIMOT tablet in 0.1 N HCl, the middle layer disintegrated rapidly within 28 seconds, allowing the floating layer to separate. The detached floating layer rose to the surface of the medium within 32 seconds, indicating a very short floating lag time. The tablet floated for more than 7 hours, confirming the efficiency of the floating layer in providing prolonged gastric retention.


Figure 17: RIMOT after gastric separation
In vitro drug release of RIMOT
At 2 h, the formulation released 29.8±0.674% of the drug. By 5 h, the release reached 66.2±1.32%, indicating almost complete release of the microsphere along with partial contribution from the colon-targeted layer. At the end of 7 h, the total percentage cumulative drug release was 75.3±0.731%, confirming sustained release from the colon-targeted tablet.
Table 16: Invitro drug release of RIMOT
|
Time (h) |
%CDR |
|
2 |
29.8±0.674 |
|
5 |
66.2±1.32 |
|
7 |
75.3±0.731 |

Figure 18: In vitro drug release of RIMOT
SUMMARY AND CONCLUSION
This study successfully developed a Rocket-Inspired Multi-site Oral Tablet (RIMOT) designed to release a drug at different regions of the gastrointestinal tract using a stage-separation concept similar to multi-stage rockets. The colon-targeted layer formulated with guar gum exhibited acceptable pre- and post-compression properties and released the drug specifically in colonic conditions, with F3 performing optimally. Loperamide-loaded microspheres prepared by ionic gelation served as the intestinal delivery system, with F3 showing the best entrapment and controlled release. The floating layer, containing HPMC and sodium bicarbonate, provided rapid buoyancy and sustained floating, ensuring gastric retention; F3 again showed the best performance. The assembled RIMOT tablet demonstrated effective layer separation, mimicking rocket stage detachment, enabling sequential drug release in the stomach, small intestine, and colon. All optimized formulations met Pharmacopeial standards and showed desirable in vitro behavior. Overall, the RIMOT system proved to be a promising platform for achieving controlled, site-specific, multi-site drug delivery and may be adaptable for various drugs requiring targeted release across different GI regions.
REFERENCES


Raeesa K. U.*, Dr. Geetha V. S., Anagha George, Rocket-Inspired Oral Tablet Formulation for Multi-Site Drug Delivery, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 649-666. https://doi.org/10.5281/zenodo.17806411
 10.5281/zenodo.17806411
10.5281/zenodo.17806411
