Crescent College of Pharmaceutical Sciences, Payangadi, Kannur, Kerala, India 670358
Inflammation is a crucial biological process that protects the body against harmful stimuli; however, its prolonged or dysregulated activation is associated with the development of various chronic diseases, including arthritis, cardiovascular complications, metabolic disorders, and neurodegenerative conditions. Although several anti-inflammatory drugs are clinically available, many of them exhibit significant limitations such as gastrointestinal toxicity, reduced selectivity, diminished long-term efficacy, and the potential for drug resistance. These challenges highlight the urgent need to explore and develop novel anti-inflammatory agents with improved safety and therapeutic profiles. Heterocyclic scaffolds remain a vital class of compounds in modern medicinal chemistry. Among these, the indole framework is recognized for its broad pharmacological relevance, demonstrating anti-inflammatory, antimicrobial, anticancer, and analgesic properties. Similarly, thiazolidine-2,4-dione (TZD) derivatives possess diverse biological activities, including antioxidant, antidiabetic, and anti-inflammatory effects. The strategic integration of these two pharmacophores into a single hybrid molecule represents a promising approach to enhance potency and achieve synergistic therapeutic benefits. Advancements in computer-aided drug design (CADD) have dramatically accelerated the discovery of new chemical entities by enabling the prediction of drug–target interactions and optimizing lead compounds with improved affinity, selectivity, and pharmacokinetic behavior. In the present study, indole-linked TZD hybrids were rationally designed using computational tools loke AutoDock Vina, synthesized through appropriate organic methodologies. The structural confirmation was done by different spectroscopic methods and evaluated for their biological activity by protein denaturation inhibition assay. The overall objective of this work is to identify potential indole-TZD hybrid molecules with enhanced anti-inflammatory activity and reduced adverse effects, thereby contributing to the development of more effective therapeutic alternatives for inflammation-related disorders.
Inflammation is a fundamental protective response that enables the body to eliminate harmful stimuli such as pathogens, damaged cells, or irritants1. Although acute inflammation is essential for host defense, dysregulated or chronic inflammation contributes to the progression of several diseases, including rheumatoid arthritis, osteoarthritis, inflammatory bowel disorders, cardiovascular diseases, and various metabolic abnormalities2. Current anti-inflammatory drugs, such as non-steroidal anti-inflammatory drugs (NSAIDs) and corticosteroids, remain the primary treatment options; however, their long-term use is limited by gastrointestinal toxicity, renal complications, cardiovascular risks, and reduced therapeutic tolerance3,4. Consequently, there is continuous interest in identifying new chemical entities with improved safety profiles and effective inflammation-modulating potential5. Heterocyclic scaffolds have gained particular importance in medicinal chemistry due to their broad pharmacological versatility and favorable drug-like characteristics6.
Thiazolidine-2,4-dione (TZD) derivatives have attracted sustained attention for their diverse biological activities, including anti-inflammatory, anti-diabetic, anticancer, and antimicrobial effects7,8. The TZD ring is known to act as a pharmacophoric motif capable of interacting with various biological targets such as cyclooxygenases, lipoxygenases, and peroxisome proliferator-activated receptors (PPARs)9,10. Structural modifications at the 5-position of TZD often enhance its biological performance, making it a promising platform for the development of synthetic anti-inflammatory agents11.
Indole is another privileged heterocyclic nucleus widely present in natural products and therapeutic agents. Its structural resemblance to tryptophan and other endogenous ligands allows indole derivatives to engage effectively with diverse biological targets12. Indole-based compounds have been reported to display anti-inflammatory, analgesic, antioxidant, antimicrobial, and anticancer activities. Owing to its planar aromatic system, hydrogen-bonding potential, and binding flexibility, the indole ring is frequently used in rational drug design to improve affinity and specificity13.
Given the complementary biological relevance of both the indole and thiazolidine-2,4-dione scaffolds, hybridization of these two pharmacophores provides an attractive strategy for generating new molecules with synergistic anti-inflammatory potential14. Molecular hybridization aims to integrate two active frameworks into a single structural architecture to achieve enhanced potency, reduced toxicity, and potentially new modes of action15. In this context, the present study focuses on the strategic design and synthesis of indole-linked thiazolidine-2,4-dione hybrids to evaluate their anti-inflammatory activity16.
To explore structure–activity relationships, two specific derivatives were synthesized featuring electron-withdrawing substituents on the indole ring: a 2-chloro-substituted compound and a trifluoro-substituted compound17. Electron-withdrawing groups are known to influence physicochemical properties such as lipophilicity, electron density distribution, and receptor-binding affinity18,19. The presence of a chloro group may enhance hydrophobic interactions within the target binding site, whereas a trifluoromethyl group often improves metabolic stability and biological potency due to its strong inductive effects20. Evaluating these substituents provides insight into how electronic factors affect the anti-inflammatory efficiency of the newly synthesized hybrids21.
The synthetic route adopted for the preparation of the indole–TZD hybrids involved standard condensation and cyclization reactions, enabling efficient linkage between the two heterocyclic systems22. Structural confirmation of the synthesized derivatives was performed through infrared (IR) spectroscopy and proton and carbon nuclear magnetic resonance (¹H and ¹³C NMR) spectroscopy23. The characteristic IR absorption peaks for the TZD carbonyl groups, indole functionalities, and other diagnostic vibrational bands confirmed the formation of the desired hybrids. NMR data further supported structural integrity through chemical shift patterns, coupling constants, and integrations consistent with the expected molecular frameworks24,25.
The anti-inflammatory activity of the synthesized compounds was assessed using in vitro evaluation models, namely the protein denaturation assay. Protein denaturation is a well-established model for screening anti-inflammatory agents because many inflammatory diseases involve protein denaturation in their pathological mechanisms29. Compounds that prevent heat-induced denaturation of proteins demonstrate their potential to mitigate inflammatory responses. Membrane stabilization is an important mechanism through which anti-inflammatory agents can reduce the release of inflammatory mediators30.
The results obtained from the tests provide comparative insights into the activity of the 2-chloro and trifluoro substituted indole–TZD hybrids, highlighting the role of electronic substituents in modulating anti-inflammatory behavior. Together, the design, synthesis, and evaluation of these hybrid molecules contribute to ongoing efforts to identify structurally novel and biologically promising anti-inflammatory drug candidates31.
REVIEW OF LITERATURE
1. Ayaz Mahmood Dar et al;2017, Molecular docking has emerged as a vital bioinformatics tool used to model and analyze the interaction between ligands and target receptors for predicting stable molecular complexes. It estimates the three-dimensional conformation of ligand–receptor adducts based on their binding affinities and energetic stability. The docking process generates multiple possible conformations, which are subsequently ranked using scoring functions within specialized software to identify the most favorable binding pose. Despite its wide applicability in structure-based drug design, molecular docking still faces several challenges, including ligand- related issues such as tautomerism and ionization, limited receptor flexibility due to the assumption of a single rigid conformation, and inaccuracies in scoring functions that can hinder precise prediction of true binding modes. Recent literature emphasizes continuous advancements in docking algorithms, scoring accuracy, and receptor modelling techniques to improve the reliability of docking-based drug discovery.
2. A. Lakshmana Rao et al; (2018) The review explores the synthesis of 3-[substituted phenylamino-methyl]-5-[(indol-3-yl)methylene]-TZD derivatives by mannich reaction with formaldehyde and aromatic amines through a conventional heat method at 120 degree celcius. Conventional heating relies on surface-to-core transfer, leading to large temperature gradients, longer processing times, and lower energy efficiency compared to modern technologies like microwave heating. Modern alternatives like microwave, radio frequency, infrared, and ohmic heating, which often provide faster, more uniform, and more energy-efficient heating.
3. Deepak pandiar et al. (2023) the study explores the anti-inflammatory activities of herbal formulation made from combination of C. nucifera and T. aestivum extract using method called protein denaturation assay. Diclofenac is used as standard in the study this method aims to avoid animal experimentation and offers a low cost ,simple and less toxic approach. The percentage inhibition of protein denaturation was taken to measure anti-inflammatory activity. And this suggest that extract could be used as a remedy for anti-inflammation.
4. Ahmed Elkamhawy et al. (2020) The study explores the thiazolidine-2,4-dione derivatives have been extensively explored as multifunctional pharmacophores exhibiting antidiabetic, anticancer, and anti-inflammatory properties . Their activity is often attributed to their ability to modulate key enzymes such as cyclooxygenase(COX), lipoxygenase (LOX), and inducible nitric oxide synthase (iNOS).Recently several studies reported synthesis of hybrid molecules integrating indole and thiazilidine-2,4-dione cores to achieve improved pharmacological activity. Indole moieties , being privileged scaffolds in medicinal chemistry, are known to enhance lipophilicity and receptor binding affinity through hhhhh stacking and hydrogen bonding interaction with target protiens. Docking studies confirms strong interaction with the cox-2 active site , supporting their anti- inflammatory mechanism of action.
5. Shankar G. Alegaon et al. (2024) Study reported the synthesis of series of heterocyclic compounds through concise three step reaction process involving knovenagel condensation at the 5th position of 2,4 thiazolidinedione ring system. The biological evaluation of these derivatives revealed promising pharmacological potential in multiple therapeutic areas. The in-vivo anti diabetic activity was evaluated using diabetes induced wistar rats , while anti inflammatory activity was studied through carrageenan and formalin induced paw edema models in rats . molecular docking studies using schr?dinger glide module demonstrated favourable binding interaction of the most potent antidiabetic derivatives with active site of PPAR-γ.Among all synthesized compounds , particular compound shows exhibit significant anti diabetic activity when compared to the standard drug pioglitazone, it also shows strong anti-inflammatory activity in the carrageenan induce paw edema model.
6. Benjamin A . Babalola et al. (2025) study explores the indole derivatives represent an important class of bioactive compounds in medicinal chemistry due to the their structural flexibility and diverse pharmacological activities. Indole derivatives exhibit wide ranging biological activities, including antifungal, antibiotic, anti-inflammatory, antioxidant, antibacterial and anticancer effects. Structural modifications such as indole-triazole and indole-thiazolidine-2,4-dione conjugates have enhanced pharmacological potency. Molecular docking and computational studies further conform their strong binding affinity towards key biological targets. Overall, indole derivatives continue to serve as promising lead structures for the development of novel therapeutic agents.
7. Lovering et al. (2004) demonstrated that the antitumor effects of NSAIDs cannot be explained solely by COX inhibition, as previously believed. Their work highlighted AKR1C3 as a significant COX-independent target. They showed that certain NSAIDs, including indomethacin and flufenamic acid, directly bind to AKR1C3 and may influence cancer-related pathways. Using high-resolution X-ray crystallography, they found that indomethacin occupies the enzyme’s active site, whereas flufenamic acid interacts both with the active site and a β-hairpin loop region. The study also reported structures with acetate in the proposed oxyanion hole, clarifying key features of the enzyme’s catalytic mechanism. These findings strengthen the idea that AKR1C3 contributes to NSAID-mediated antineoplastic activity and provide a structural foundation for designing cancer therapies with fewer COX-related side effects.
2. MATERIALS AND METHODS
2.1. Materials
The chemicals and reagents were procured from Sigma Aldrich, Alchemy lab solutions, Edapally Kochi. Melting point was determined by open capillary method. IR spectra were recorded on Bruker FT-IR (Shimadzu 8201 PC) spectrophotometers (Alshifa collage of pharmacy, Perithalmanna) and values are expressed in cm-1. 1H NMR and 13C-NMR spectra were recorded on Bruker Avance-500 FTNMR spectrometer (JSS College of Pharmacy, SS Nagar, Mysore, Karnataka ) at 500MHz and the chemical shifts are reported in parts per million (δ value), taking TMS (δ 0 ppm for 1H NMR) as the internal standard.
2.2 IN-SILICO STUDY
All computational analysis was carried out on a Windows 10 Pro OS platform on a Desktop with an Intel(R) Pentium(R) CPU J3710 @ 1.60GHz and 4 GB RAM.
Physiochemical properties
Molinspiration Molecular Viewer allows the visualization of molecules which is encoded as SMILES or SD file for the calculation of important molecular description (Log P, Polar surface area, Number of hydrogen bond donors, Number of hydrogen bond acceptors etc.) as well as prediction of bioactivity score of important drug targets32.
2.3 PHARMACOKINETIC STUDY BY SWISSADME
SwissADME is a web tool giving free access into physiochemical properties (Molecular weight, Molar refractivity, Polar surface area) pharmacokinetics (substrate or non-substrate of P-gp, CYP inhibition), drug-likeness of potent molecule. It also produces predictive models such as boiled egg allows the evaluation of human intestinal absorption (HIA) and brain penetration (BBB) of drug molecule. The white region is for high probability of passive absorption by the GI tract and yolk region is for high probability of brain penetration33.
Lipinski’s rule of five
Lipinski’s rule is a tool to access ADME properties and predict drug-likeness. The rule shows the following extensions:
2.4 IN-SILICO TOXICITY PREDICTION – PROTOX.3.0
In silico toxicity prediction is done using PROTOX.3.0 Property Explorer. It is a free software available for access in the Organic Chemistry Portal. Using this prediction tool, mutagenicity, tumerogenicity, skin irritation and reproductive effects can be calculated. The prediction properties relies on a precompiled set of structure fragment that gives rises to toxicity alerts in case they are encountered in the structure currently drawn. These fragment lists is created by rigorously shredding all compounds in the data base known to be active in a certain toxicity class. During the shredding any molecule is first cut at every rotatable bonds leading to a set of core fragments35.
PASS Online
Prediction of Activity Spectra for Substances (PASS) is a computer program that allows to estimate the probable profile of biological activity of a drug- like organic compound based on its structural formula36. It provides an output information as a list of predicted types of activity with estimated probability for each type of activity ‘to be active’ Pa and ‘to be inactive’ Pi, which may vary from zero to one.
2.5 MOLECULAR DOCKING
Docking of small molecules and compounds into the binding site of receptor and estimating the binding affinity of complex is consider to the important part of structure based drug design38. Molecular docking is achieved by Autodock Vina. The 3D crystallographic structure of proteins are uncovered from protein data bank (PDB ID-1S2A).
Autodock Vina is an open source program offering a complete molecular viewer and graphical support for all the steps inevitable for set up and docking analysis. PyMOL produce a high quality 3D image of protein as well as its visualization. PyRx is for docking analysis39,40.
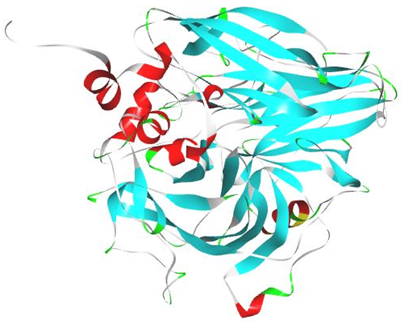
Figure no 1: Protein (PDB ID: 1S2A)
Protein preparation
Structure downloaded from PDB database is unsuitable for docking studies. PyMOL produce high quality 3D image of these protein. The structure should clear up with water molecules (HOH), small molecules and detergents (DSN). It is achieved by inserting various commands like “remove<>resn<> HOH/DSN” (for water molecules or detergents). Hydrogen atom should be added to proteins structure41.
Ligand preparation
The 2D chemical structure of ligands are drawn using ACD Lab Chemsketch ver 12.0 or Chemdraw Ultra 12.0 and generated smiles notation. This smile notation is being converted into 3D PDB format with the help of freely accessible Online SMILES Translator42.
Docking using Autodock Vina
Docking is performed in PyRX where both the derivative and receptor are loaded in navigation plane. Then the protein is converted into macromolecule and derivative into ligand molecule. After the preparation, click on Autodock Vina Wizard start button and adjust the grid size. After the completion of process, docking value is expressed in terms of binding affinity (Kcal/mol) with RMSD upper bound and lower bound value. Autodock Vina convert PDB to PDBQT which is followed by additional step by adding polar contacts to find out the types of amino acid interactions during ligand-receptor binding43.
2.6 CHEMISTRY
1. General procedure for the Synthesis of 5-[(indol-3-yl)methylene]-thiazolidine-2,4-dione
Piperidine (0.4 mL) was added to a solution of thiazolidine- 2,4-dione (1.17 g, 0.01 mol) and indole-3-carboxaldehyde (1.45 g, 0.01 mol) in toluene (8mL) The reaction mixture was placed in Raga’s scientific microwave synthesis reaction vessel, which was connected with a water condenser. The reaction mixture was irradiated at 350W for about 8 min at 120?
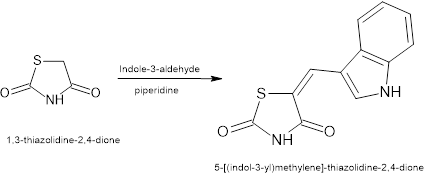
Figure no 2: Syntesis of 5-[(indol-3-yl)methylene]-thiazolidine-2,4- dione
2. General procedure for the synthesis of 3-[(substitutedphenylamino)methyl]-5-[(indol-3-yl)methylene]-thiazolidine-2,4-dione
To a solution of 5-[(indol-3-yl)methylene]-thiazolidine- 2,4-dione (2) (0.005 mol) in of DMF(3 mL), formaldehyde (0.01 mol) was added and the reaction mixture stirred for about 20 to 30 min at room temperature. The solution of aryl amine (0.005 mol) in DMF and catalytic amount of conc. HCl (3e5 drops) were added to the above reaction mixture. The resulting mixture was placed in Raga’s scientific microwave synthesis reaction vessel and was irradiated at 420W for about 8 to12 min at 120 ?
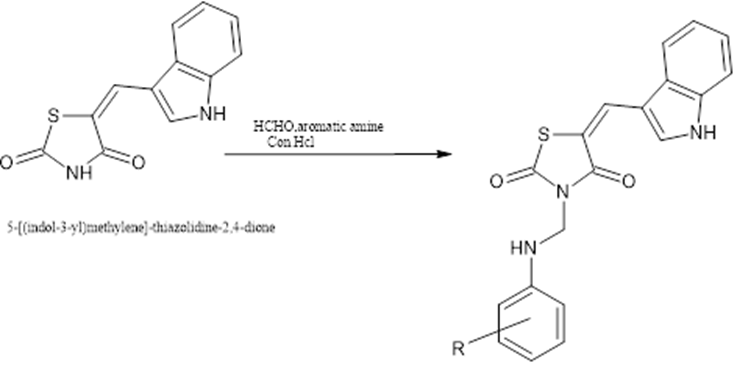
Figure no 3: synthesis of 3-[(substitutedphenylamino)methyl]-5-[(indol-3-yl)methylene]-thiazolidine-2,4-dione
2.7 REACTANTS
1,3-thiazolidine-2,4-dione
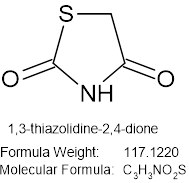
Figure no 4: 1,3-thiazolidine-2,4-dione
5-[(indol-3-yl)methylene]-thiazolidine-2,4-dione
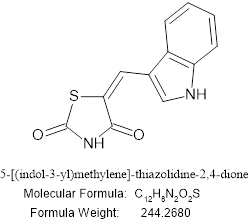
Figure no 5: 5-[(indol-3-yl)methylene]-thiazolidine-2,4-dione
Aniline derivatives
(a) (b) (c) (d)
Figure no 6: (a)aniline, (b)2-chloroaniline, (c)4-(trifluromethoxy)aniline, (d)2,4-dichloroaniline
2.8 STRUCTURE OF SYNTHESIZED COMPOUNDS
TZD1
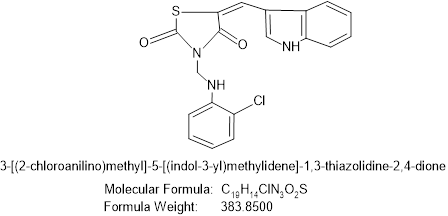
Figure no 7: 3-[2-chloro-N-methylaniline]5-[(indol-3-yl)methylene]-thiazolidine-2,4-dione.
TZD 4
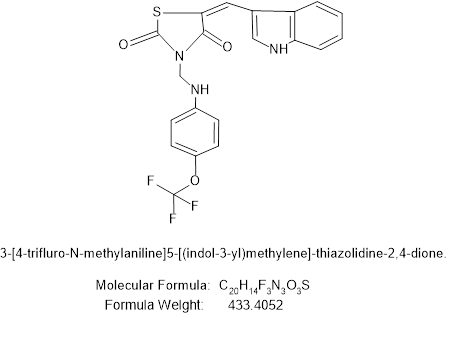
Figure no 8: 3-[4-trifluoromethoxy-N-methylaniline]5-[(indol-3-yl)methylene]-thiazolidine-2,4-dione.
2.9.CHARACTERIZATION
Melting point
The melting point of the synthesized compound was determined by open capillary tube
method. The temperature at which the compound starts losing its crystallinity and changes from
solid to liquid form was found and recorded.
Thin layer chromatography
The reactants and products were dissolved in ethanol. It was spotted on the TLC plate. A single principal spot for the product and the absence of secondary spots for parent compounds and
intermediates confirmed the purity of the product. Stationary phase: pre-coated silica gel GF
using appropriate mobile phase was used. The spots were detected in a UV chamber45.
IR spectrometry
Infrared spectroscopy is one of most commonly used spectroscopic technique for
identification of functional groups in molecules. IR spectroscopy is an important tool in the
structural elucidation of organic compounds. In IR spectroscopy finger print region is used to
compare the two compounds. Infrared spectrum shows percentage transmittance versus
frequency expressed as wave numbers.
NMR spectroscopy
Nuclear Magnetic Resonance (NMR) spectroscopy is an important analytical technique used in the structural elucidation of organic molecules. It involves the interaction of the electromagnetic radiation and the proton of a nucleus of an atom when placed under an externally applied static magnetic field. NMR spectra provide the detailed information about a molecule’s structure. The chemical shift is used to predict the number of protons with refers to TMS as standard. The NMR spectra is recorded on 300 MHz BRUKER advance III NMR spectrometer. DMSO is used as a solvent46.
2.10 IN-VITRO ANTI-INFLAMMATORY ASSAYS
a. Protein Denaturation Method
Principle:
Protein denaturation has been well correlated with the occurrence of the inflammatory response and leads to various inflammatory diseases including arthritis. Denaturation of the proteins is a condition when the unique three-dimensional structure of a protein is exposed to changes by breaking of many of the weak linkages, or bonds (e.g., hydrogen bonds), within a protein molecule that are responsible for the highly ordered structure of the protein in its natural (native) state. The protein denaturation assay is based on the idea that substances with anti-inflammatory qualities may be able to stabilize protein structures and prevent denaturation, which is frequently linked to inflammation and tissue damage.
Procedure:
The reaction mixture (0.5 ml) consisted of 0.4 ml bovine serum albumin (3% aqueous solution) and varying concentration of test sample. The samples were incubated at 37ºC for 20 minutes and 2.5 ml phosphate buffered saline (pH 6.3) was added to each tube and then heated at 80ºC for 10 minutes. The absorbance was measured using spectrophotometer at 660nm47.
The percentage inhibition of protein denaturation was calculated as follows:
Percentage inhibition =
Absorbance of Control -Absorbance of test ×100Absorbance of control
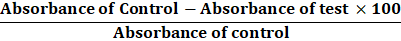
3. RESULTS AND DISCUSSION
3.1 CHEMISTRY
The synthesized compound (TZD4) were obtained good yields (68–85%). Spectroscopic
data confirmed the proposed structures.
IR spectra :
Yellow powder (DMF); MP = 180-185 ? C; Yield 64.3% w/w; IR [KBr V cm "]: 3241 (-NH-), 3462(-NH-), 1687 (C=0), 1773.35(C=0), 1303 (C-N), 2931 (?-?), 3059 (=?-?),606 (C-S), 1138 (C-F).
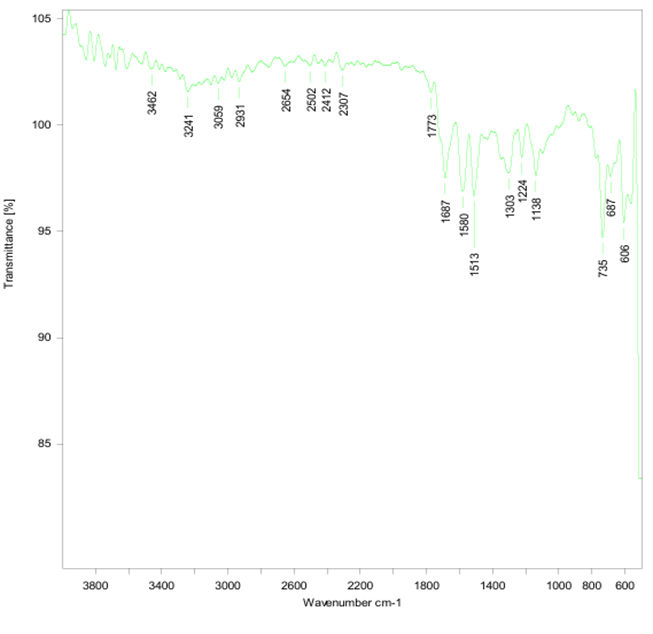
Figure no 9: IR spectrum of TZD4
The structure of obtained compounds was confirmed by spectroscopic techniques like FTIR,. IR Spectrum of synthesized compounds showed the single characteristic peak of secondary amine stretching in the range 3330-3310 cm-1which confirms the presence of secondary amine in the synthesized compounds.
1H NMR :
1H-NMR- (400 MHz, DMSO-D6) δ 12.28 (s, 1H), 12.12 (s, 1H), 8.05 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 2.9 Hz, 1H), 7.50 (d, J = 7.9 Hz, 2H), 7.27-7.19 (m, 4H)
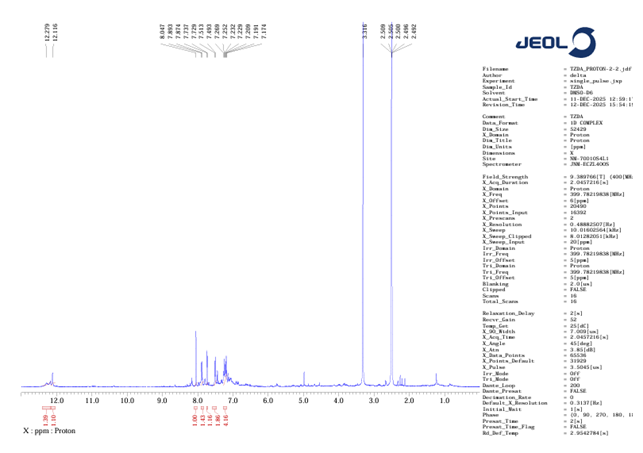
Figure no 10: 1H NMR of TZD4
The 'H NMR spectrum showed characteristic NH resonances of the indole and thiazolidinedione moieties at 12.27 and 12.11 ppm, respectively, along with aromatic proton signals between & 7.17-8.05 ppm and a singlet corresponding to the olefinic methine proton of the methylene linkage. A singlet at ? ~3.10 ppm confirmed the presence of the N-methyl group.
13C NMR :
13C-NMR- (101 MHz, DMSO-D6) δ 167.8, 167.5, 136.2, 128.6, 126.8, 124.4, 123.0, 121.0, 118.3, 116.4, 112.4, 110.5, 40.1, 39.9, 39.7, 39.5, 39.3, 39.1, 38.9
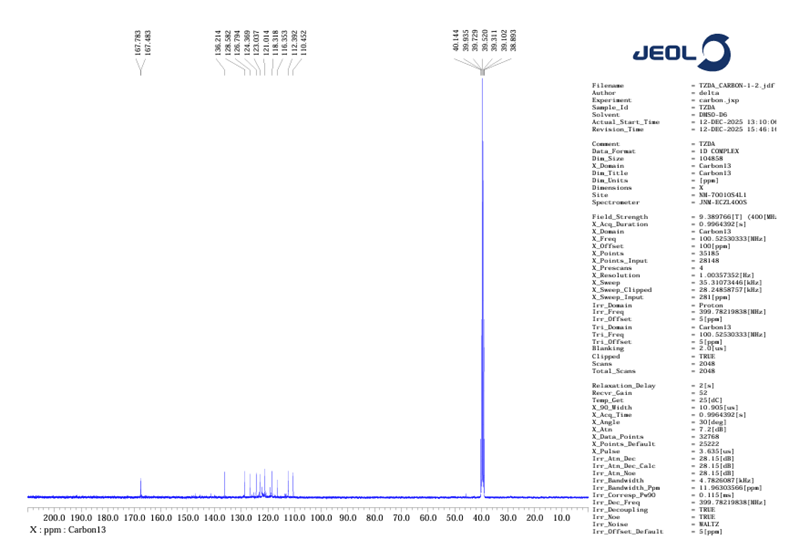
Figure no 11: 13C NMR of TZD4
The 13C NMR spectrum displayed two carbonyl carbons of the thiazolidinedione ring at ~167 ppm and aromatic carbons in the expected region.
19F NMR :
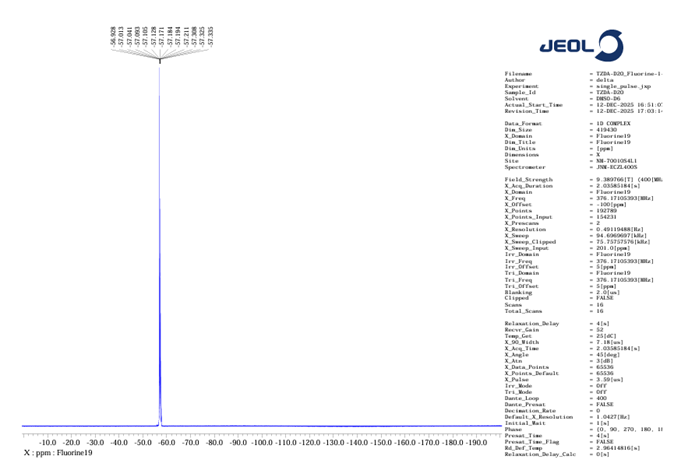
Figure no 12: 19F NMR of TZD4
The 19F NMR spectrum exhibited a single sharp resonance at -57 ppm, confirming the presence of a trifluoromethoxy group.
These spectral data are in full agreement with the proposed structure
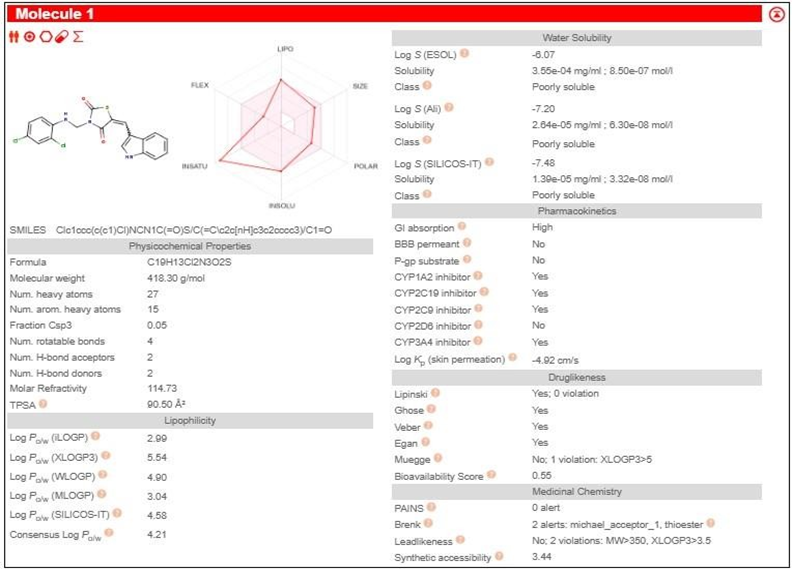
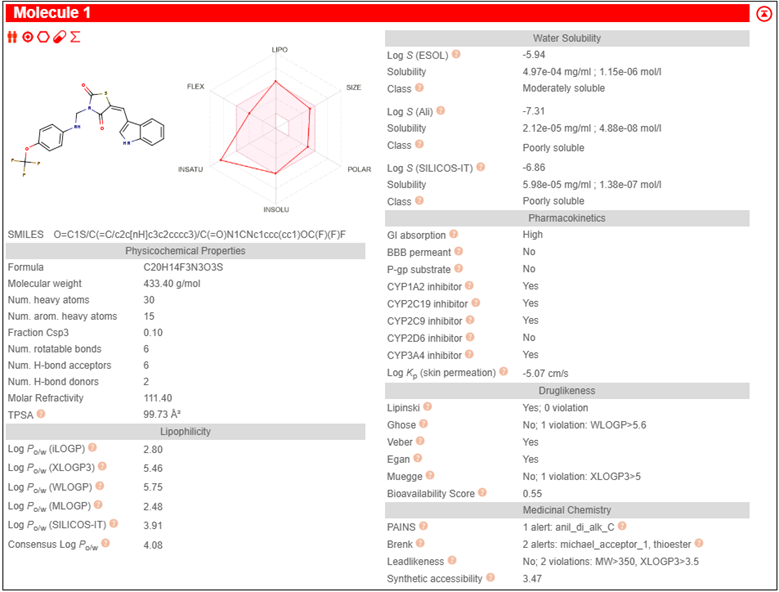
The pharmacokinetic parameters of the derivatives were determined using Molinspiration Online software. Among these parameters, the compounds that complied with Lipinski's rule of five were chosen for further docking studies.
Table 1: Analysis of Lipinski’s rule of five by Molinspiration
|
COMPOUND ID |
MOLECULAR PROPERTIES |
|
TZD1 |
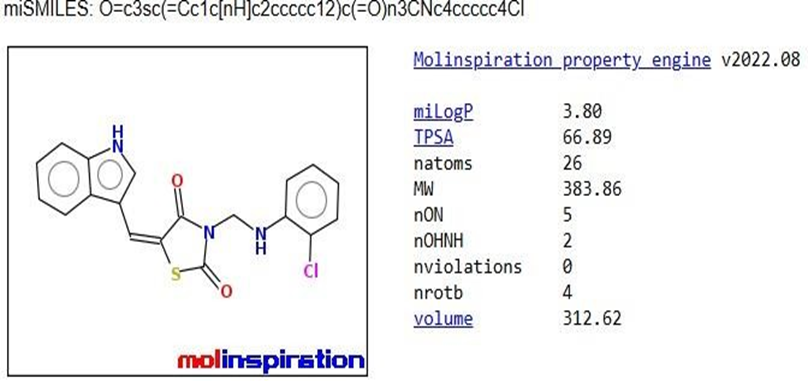
|
|
TZD2 |
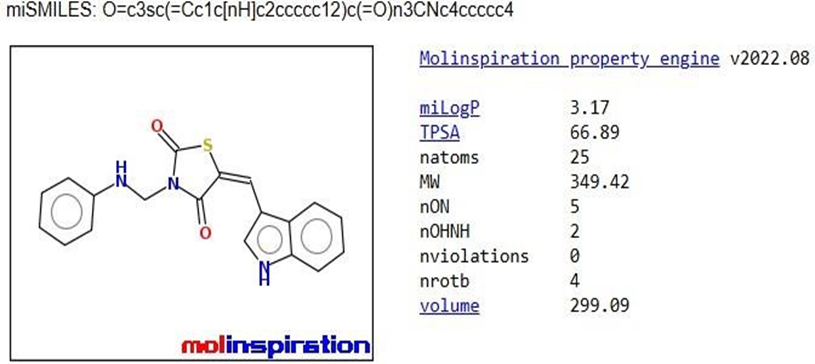
|
|
TZD3 |
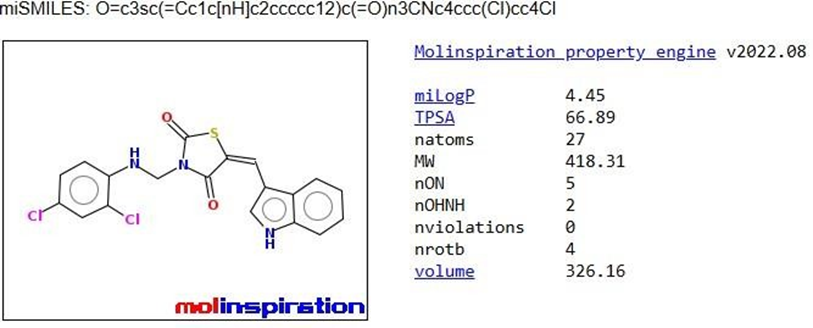
|
|
TZD4 |
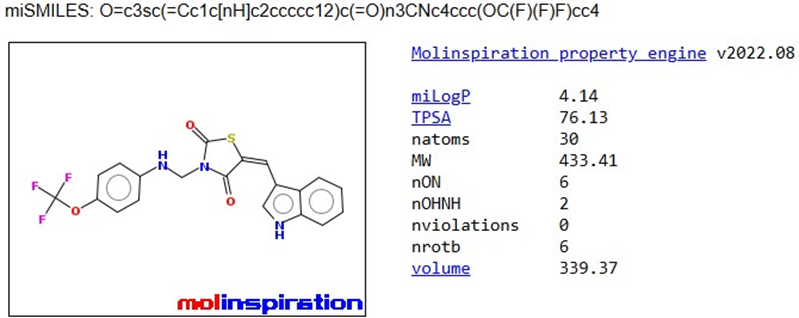
|
3.3 PHARMACOKINETIC STUDY SWISSADME
Table 2: Pharmacokinetic study by SwissADME
|
COMPOUND |
PHARMACOKINETIC PROPERTIES |
|
TZD1 |
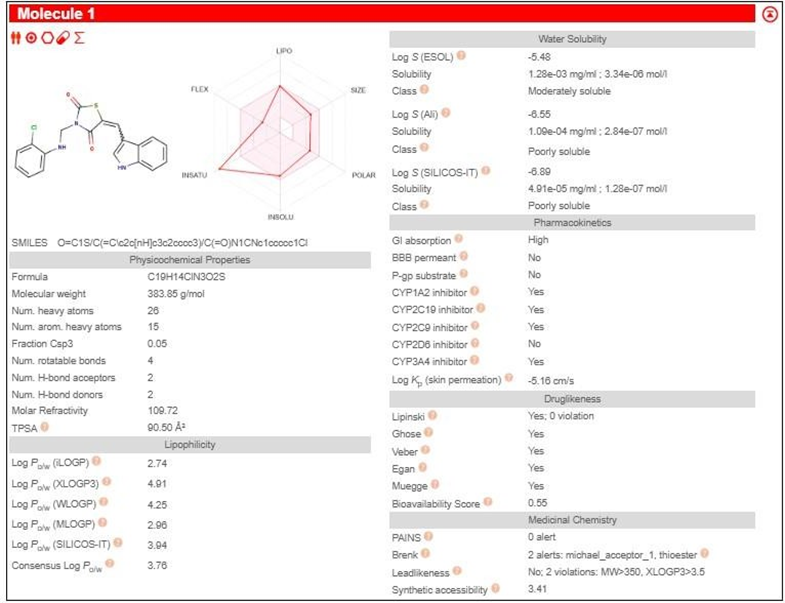
|
|
TZD2 |
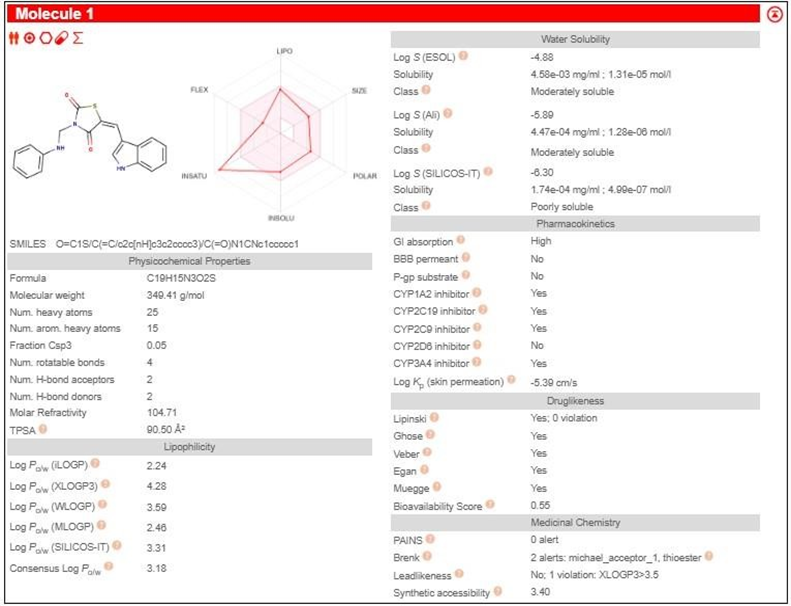
|
|
TZD3
|
|
|
TZD4 |
|
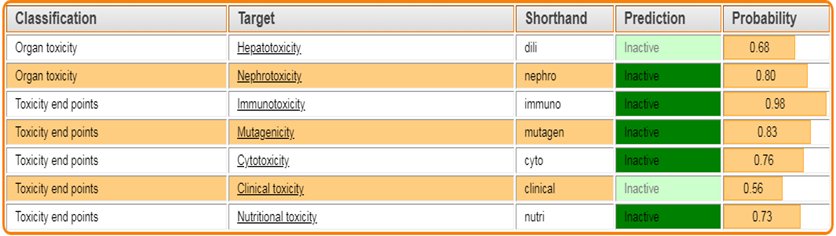
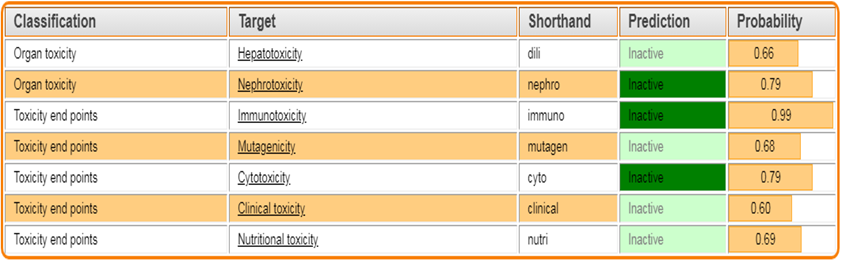
3.3 IN-SILICO TOXICITY PREDICTION – Protox.3.0:
The Protox 3.0 software is used to predict whether the designed molecules are toxic
Toxicity classes are defined according to the globally harmonized system of classification of labeling of chemicals (GHS). LD50 values are given in [mg/kg):
Table 3: Toxicity prediction by PROTOX.3.0
|
COMPOUND ID |
PROTOX TOXICITY |
|
TZD1 |
|
|
TZD2 |
|
|
TZD3 |
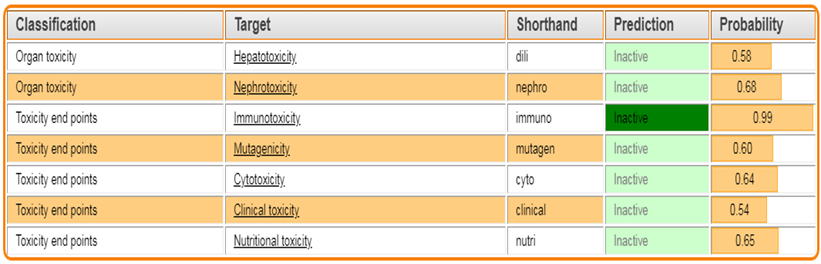
|
|
TZD4 |
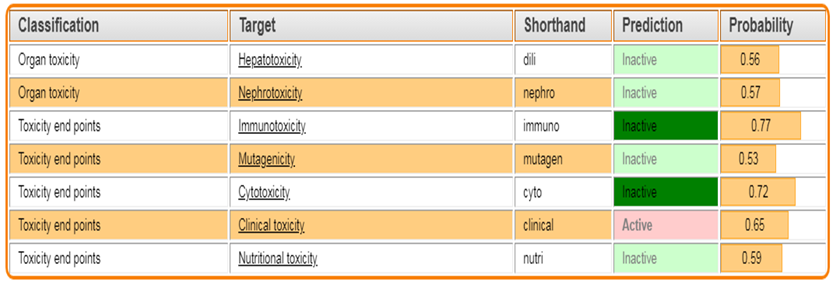
|
3.4 DOCKIMG STUDIES
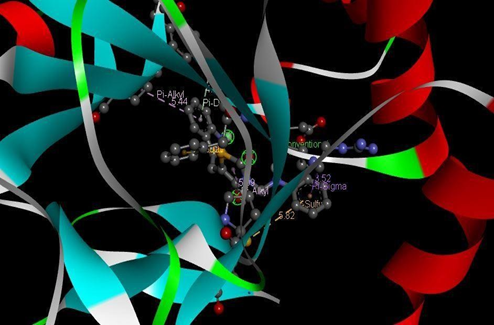
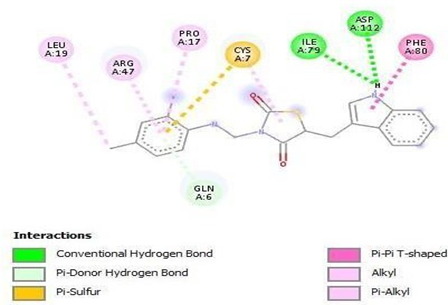
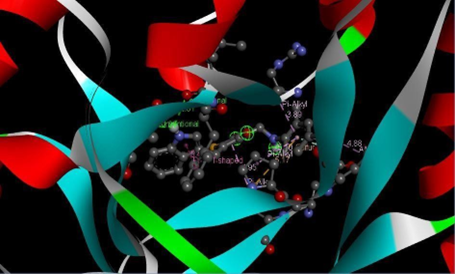
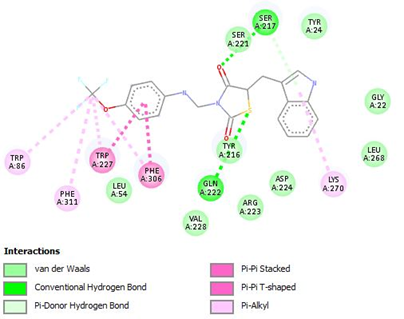
The designed molecules underwent docking against selected protein(PDB ID:1S2A) and the compounds were filtered and sorted based on their respective docking score. The most favorable and stable pose was selected based on the docking score and multiple interactions observed. Upon comparing the binding structures of the three molecules, it was found that the thiazolidinedione rings played a crucial role in determining the binding affinity within the binding pocket. Among the docked molecules, TZD1 exhibited the highest binding affinity towards the protein, while TZD3 demonstrated the lowest binding affinity.
Table 4: Docking Results
|
Sr. No |
Compound |
Structure |
Docking score |
Interacting residue |
|
1 |
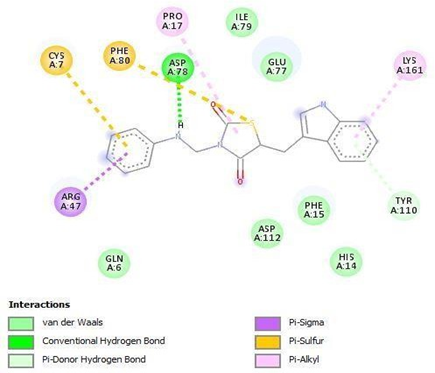
TZD1 |
|
-10.5 |
HIS A: 304 ASN A: 302 PRO A: 318 |
|
2 |
TZD2 |
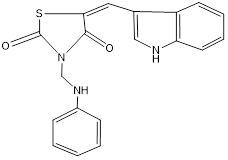
|
-10.1 |
ASN A: 302 TYR A:319 ASP A: 300 |
|
3 |
TZD3 |
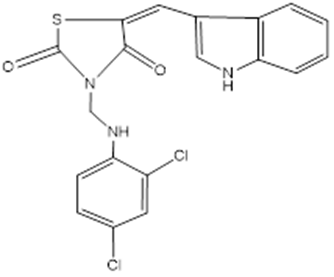
|
-8 |
HIS A:304 SER A:320 TYR A:319 ASP A:300 PRO A:318 ASN A:302 |
|
4 |
TZD4 |
|
11.3 |
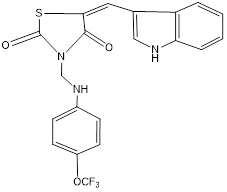
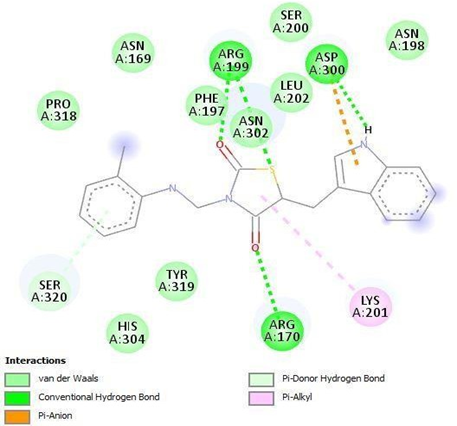
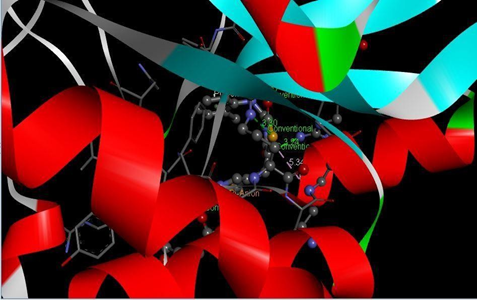
HIS A:304 SER A:320 TYR A:319 ASP A:300 PRO A:318 ASN A:302 |
(a) (b)
Figure 13: Binding interaction of TZD1(a) and binding pattern of TZD1 in binding pocket (b)
(a) (b)
Figure 14: Binding interaction of TZD2(a) and binding pattern of TZD2 in binding pocket (b)
(a) (b)
Figure 15: Binding interaction of TZD3 (a) and binding pattern of TZD3 in binding pocket (b)
(a) (b)
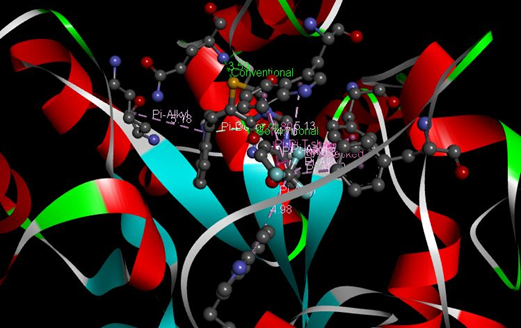
Figure 16: Binding interaction of TZD4 (a) and binding pattern of TZD4 in binding pocket (b)
3.4 BIOLOGICAL EVALUATION
Protein denaturation inhibition assay
The compound with the highest docking score (TZD1) was selected for in-vitro studies. To assess its anti-inflammatory activity protein denaturation inhibition assay was employed. The protein denaturation assay is based on the idea that substances with anti-inflammatory qualities may be able to stabilize protein structures and prevent denaturation, which is frequently linked to inflammation and tissue damage. The reaction mixture (0.5 ml) consisted of 0.4 ml bovine serum albumin (3% aqueous solution) and varying concentration of test sample. The samples were incubated at 37ºC for 20 minutes and 2.5 ml phosphate buffered saline (pH 6.3) was added to each tube and then heated at 80ºC for 10 minutes. The absorbance was measured using spectrophotometer at 660nm.
The percentage inhibition of protein denaturation was calculated as follows:
Percentage inhibition =
Absorbance of Control -Absorbance of test ×100
Absorbance of control
Table 5: Assay result and percentage inhibition of Diclofenac
|
Standard |
Concentration (μg/ml) |
Absorbance at 660 nm |
Percentage Inhibition (%) |
|
Control |
- |
0.787 |
|
|
Diclofenac |
6.25 |
0.725 |
7.88 |
|
12.5 |
0.661 |
16.01 |
|
|
25 |
0.545 |
30.75 |
|
|
50 |
0.368 |
53.24 |
|
|
100 |
0.125 |
84.12 |
|
|
IC 50 |
|
49.68μg/ml |
Table 6: Assay result and percentage inhibition of TZD4
|
Sample code |
Concentration (μg/ml) |
Absorbance at 660 nm |
Percentage Inhibition (%) |
|
Control |
- |
0.782 |
- |
|
TZD4 |
6.25 |
0.771 |
1.41 |
|
12.5 |
0.714 |
8.70 |
|
|
25 |
0.586 |
25.06 |
|
|
50 |
0.488 |
37.60 |
|
|
100 |
0.328 |
58.06 |
|
|
IC 50 |
|
80.75 μg/ml |
The test sample showed concentration-dependent inhibition of protein denaturation. At 100 µg/ml, sample exhibited 58.06% inhibition, whereas the standard drug Diclofenac showed 84.12% inhibition at the same concentration. The IC?? value of sample (80.75 µg/ml) is higher than that of Diclofenac (49.68 µg/ml), indicating moderate anti-inflammatory activity when compared to the standard.


(a) (b)
Figure 17: The turbidity of each tube indicates the extent of protein denaturation of different concentration of (a)standard and(b)sample.
4. CONCLUSION
A Series of 3-substituted-5-[(indol-3-yl)methylene]-thiazolidine-2,4-dione derivatives were developed by incorporating different aromatic amines using computational methods. Compounds with highest docking score were synthesized using conventional method. The synthesized compounds were characterized physically and most of the compounds were characterized spectrally by FT-IR, 1H-NMR, 13C-NMR.In vitro anti-inflammatory activity is evaluated using protein denaturation inhibition assay. It was found out that compound TZD4 exhibited significant anti-inflammatory compared to other compounds. The molecular docking studies also revealed that compound TZD4 exhibited highest binding affinity at AKR1C3 receptor protein. The results indicated that the novel synthesized compounds is beneficial for anti-inflammatory activity.
5. ACKNOWLEDGEMENT
It gives me immense pleasure to express my deepest gratitude to the college management and guide Dr. Sreena K, Professor and HOD Department of Pharmaceutical Chemistry, Crescent College of Pharmaceutical Sciences, Payangadi for providing the facilities for the successful completion of my project work.
6. CONFLICT OF INTEREST
We declare that we have no conflict of interest.
REFERENCES







Sreena K, Ashique P, Muhammed Shafah K P C, Thabsseera K P, Shaharban T K, Thejas T, Abid T P, Strategic Design, Synthesis and Biological Evaluation of Indole Linked Thiazolidine 2,4-Dione Hybrids as Potential Anti-Inflammatory Agent, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 4030-4052. https://doi.org/10.5281/zenodo.18096343
 10.5281/zenodo.18096343
10.5281/zenodo.18096343
