
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Rajarambapu College of Pharmacy, Kasegaon, A/P: Kasegaon Tal: Walwa Dist.: Sangli 415404 Maharashtra India.
Oxadiazole derivatives have emerged as a vital class of heterocyclic compounds in medicinal chemistry due to their diverse pharmacological activities and favorable drug-like properties. The present study focuses on the design, synthesis, in-silico analysis, and pharmacological evaluation of a series of novel substituted oxadiazole derivatives with the objective of identifying promising candidates for future therapeutic development. The structures of the synthesized derivatives were confirmed using spectral techniques such as FT-IR, ¹H-NMR, and mass spectrometry, ensuring purity and structural integrity. In-silico studies were performed to predict the biological behavior of the synthesized compounds, focusing on their molecular docking interactions with selected protein targets known to play key roles in microbial infection and inflammation. The docking results indicated strong binding affinities and stable interactions between several oxadiazole derivatives and the active sites of the target enzymes, suggesting potential for antimicrobial and anti-inflammatory activities.
In the present study, a series of substituted 1,3,4-oxadiazoles were synthesized using microwave-assisted methods and evaluated for their antibacterial and antifungal activities. Microwave synthesis proved to be highly efficient, offering significantly higher yields in a shorter time compared to conventional methods. The synthesized compounds were recrystallized using various solvents, including ethanol, petroleum ether, n-hexane, methanol, butanol, and acetone, through a slow evaporation technique. Among these solvents, ethanol and acetone produced the most well-defined crystal structures, typically forming needle-like, cross-shaped, and clustered crystals. This is particularly important, as polymorphism can greatly influence the bioavailability of drugs—especially those with poor water solubility. The structural integrity of the synthesized compounds was confirmed by satisfactory results from IR, ¹H NMR, and mass spectroscopic analyses.
1,3,4-oxadiazole- An important Heterocycle16-19: -
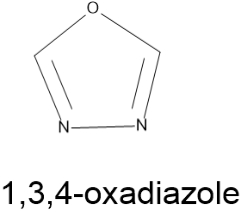
The five-membered oxadiazole nucleus is present in heterocyclic compounds with a wide range of health benefits. Oxadiazoles are cyclic compounds that include one oxygen and two nitrogen atoms in a five-membered ring. Oxadiazole is considered to be created by replacing two of the methane (-CH=) groups with two of the nitrogen (-N=) groups of the pyridine type in furan. Compounds with the 1,3,4-oxadiazole nucleus exhibit unique anti-inflammatory, analgesic, antibacterial, anticancer, anticonvulsant, anthelmintic, herbicidal, anti-mycobacterial, antioxidant, and anti-hepatitis B virus properties. Oxadiazole is a heterocyclic aromatic compound with five components, including one oxygen and two nitrogen atoms. In medicinal chemistry, they have also been studied as bioisosteres for carboxylic acids, esters, and carboxamides. It comes in a variety of isomeric forms, including 1,2,3, 1,2,4, 1,2,5, and 1,3,4-oxadiazole.
Reaction scheme:
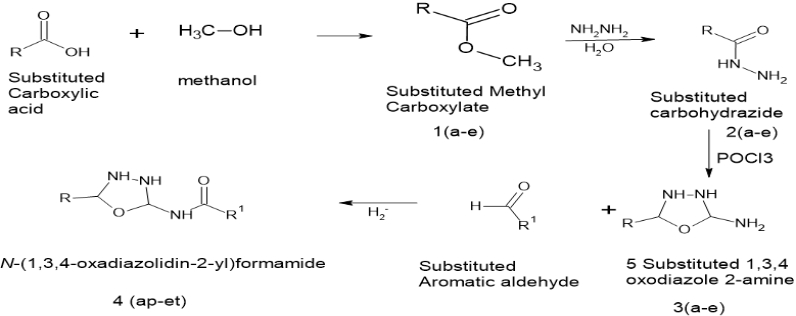
Where,
R=
R1=
(a)- Benzaldehyde
(b)Formaldehyde
(c) 4 Choro benzaldehyde
(d) 4 Dimethyl amine benzaldehyde
(e) 3 Nitrobenzaldehyde
Synthesis:
Synthesis by conventional method:
Table No. 5.1.1 Physicochemical data of 5 substituted 1,3,4 oxadiazolidin -2 yl formamide by Conventional and microwave methods
|
Sr. No. |
Com-pound code |
Molecular formula |
Molecular weight g/mol |
Melting Point °C |
Percentage yield % |
Rf Value |
Mobile phase |
|
|
Conven-tional |
Micro-wave |
|||||||
|
1 |
3a |
C16H11N5O9 |
417.29 |
120-124 |
88 |
90 |
0.74 |
Ethyl acetate :Ethanol (8:2) |
|
2 |
3b |
C16H11ClN4O7 |
406.74 |
130-134 |
84 |
80 |
0.80 |
Ethyl acetate :Ethanol (8:2) |
|
3 |
3c |
C16H12N4O7 |
372.29 |
122-128 |
74 |
88 |
0.66 |
Ethyl acetate :Ethanol (8:2) |
|
4 |
3d |
C18H17N5O7 |
415.36 |
120-126 |
86 |
92 |
0.82 |
Ethyl acetate :Ethanol (8:2) |
|
5 |
3e |
C11H8N4O7 |
324.21 |
165.5 –167.5 |
79 |
88 |
0.76 |
Ethyl acetate :Ethanol (8:2) |
|
6 |
4a |
C16H11Cl2N3O5 |
396.18 |
240–245 |
86 |
94 |
0.77 |
Ethyl acetate :Ethanol (8:2) |
|
7 |
4b |
C16H11ClN4O7 |
406.74 |
122-126 |
78 |
85 |
0.81 |
Ethyl acetate :Ethanol (8:2) |
|
8 |
4c |
C18H17ClN4O5 |
420.81 |
130-138 |
69 |
78 |
0.84 |
Ethyl acetate :Ethanol (8:2) |
|
9 |
4d |
C11H8ClN3O5 |
297.65 |
128-134 |
74 |
88 |
0.76 |
Ethyl acetate :Ethanol (8:2) |
|
10 |
4e |
C16H12ClN3O5 |
361.74 |
331–333 |
70 |
80 |
0.63 |
Ethyl acetate :Ethanol (8:2) |
Antibacterial Activity Docking Result
When compared to other compounds, compounds 3a through 4e and compound 4a had the lowest dock scores (-62.60). When we compared the results of the molecule to the literature, this docking score indicated that the suggested compounds had a good binding affinity for hinding to receptors (PDB Code-4AA7). In the binding pocket, the suggested compounds all take on a very similar conformation, displaying van der Waals interactions with the ASP5SA, ARG56A, and TYR67A amino acids as well as hydrogen bond interactions with the ARG56A amino acid. A 2D image is used to depict it (figno.6.2.4.2). 4a chemical and receptor overlay in the figure. It was discovered that the popular drug ciprofloxacin had a dock score of -65.60.
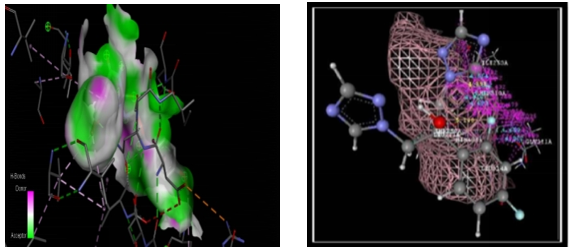
Compound code 4a Standard (Ciprofloxacin)
Fig. no. 6.2.4.1 Docking poses of compound code 4a and standard drug

Compound code 4a Standard (Ciprofloxacin)
Fig. no. 6.2.4.2: 2D representation of Docking poses of compound code 4a and standard drug
Table 6.2.4.2: Data for interaction of compound code 4a with amino acid
|
Amino acid |
Atom of ligand |
Distance |
Interaction type |
|
TYR279B |
1C |
3.991 |
AROMATIC_INTERACTION |
|
ARG56A |
956H |
2.982 |
VDW_ INTERACTION |
|
OH507C |
17C |
2.937 |
VDW_ INTERACTION |
|
ARG56A |
957H |
2.534 |
HYDROGENBOND_ INTERACTION |
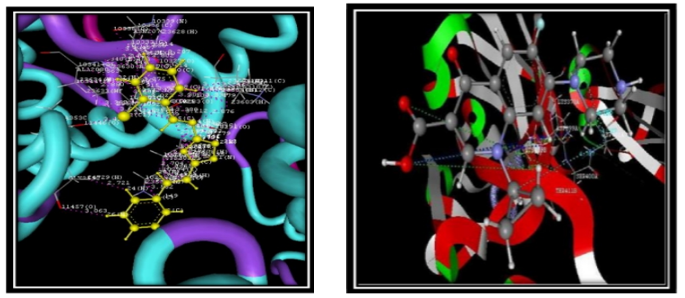
Compound code 4a Standard (Ciprofloxacin)
Fig. no. 6.2.4.3: Superimpose image representation of Docking poses of compound code 4a and standard drug
Anti-Fungal Activity Docking Result
Compounds 3a through 4e have dock scores ranging from - 48.121, with compound 3a having the minimum dock value compared to the remaining compounds. This docking score showed that the developed compounds have a strong binding affinity for binding to the receptor (PDB Code-1kZN) when we compared the results of compound 3a to the literature. The optimal position derived from the docking findings is shown in figure no.6.2.4.4. At the binding pocket, every proposed molecule takes on a fairly similar shape. This includes hydrogen bonding with the amino acid of TYR80A, vanderwal interaction binding LEU83A, hydrophobic interaction binding LEU83A, and aromatic interaction showing with the amino acid of HIS72A. This is depicted in the 2D representation diagram (fig. 6.2.4.5). Superimpose the 3a compound picture with the receptor depicted in the diagram (fig. 6.2.4.6).
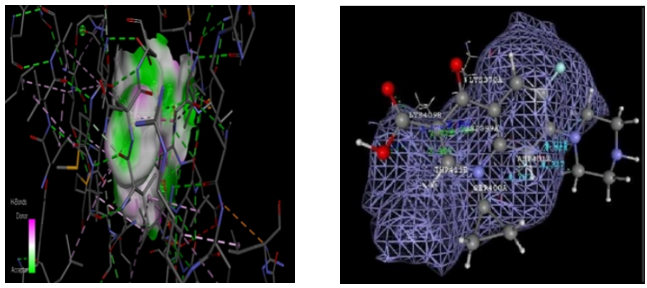
Compound code 3a Standard (Fluconazole)
Fig. no. 6.2.4.4: Docking poses of compound code 3a and standard drug
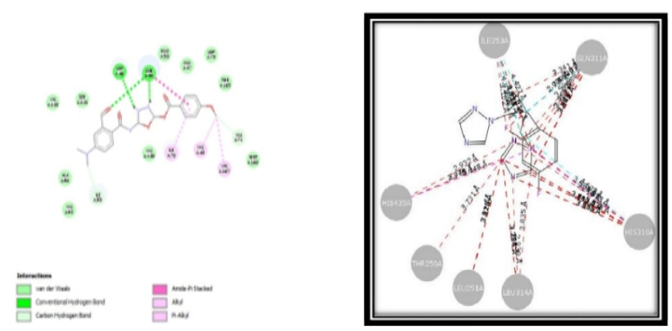
Compound code 3a Standard (Fluconazole)
Fig. no. 6.2.4.5: 2D representation of Docking poses of compound code 3a and standard drug
Table 6.2.4.4: Data for interaction of compound code 3a with amino acid
|
Amino acid |
Atom of ligand |
Distance |
Interaction type |
|
TYR80A |
9O |
1.690 |
HYDROGENBOND_ INTERACTION |
|
TYR80A |
15N |
3.750 |
VDW_ INTERACTION |
|
HIS72A |
8C |
4.310 |
AROMATIC_ INTERACTION |
|
LEU83A |
17C |
3.408 |
VDW_ INTERACTION |

Compound code 3a Standard (Ciprofloxacin)
Fig. no. 6.2.4.6: Superimpose image representation of Docking poses of compound code 3a and standard drug
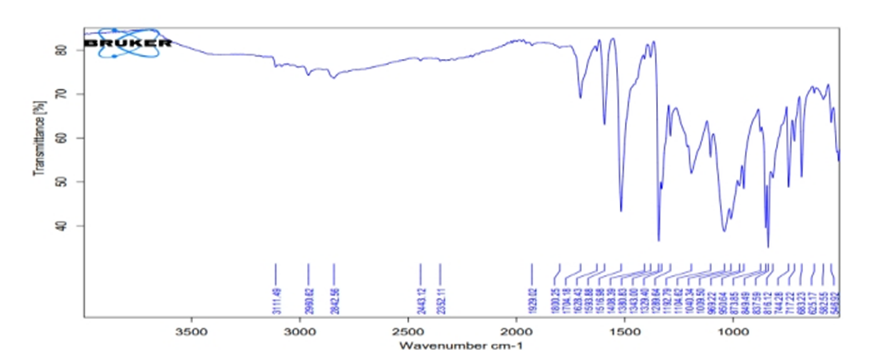
Graph No.5.2.1: IR Spectrum of 3a 5-(2 -formyl-4-nitrobenzamido)-1,3,4-oxadiaazolidin-2-yl-4-nitrobenzoate
Table. No.5.2.1: IR Spectrum of 3a 5-(2 -formyl-4-nitrobenzamido)-1,3,4-oxadiaazolidin-2-yl-4-nitrobenzoate
|
Sr. No. |
Assignment |
Functional groups |
Wavenumber (cm?¹) |
|
1 |
C-H Stretching |
Aromatic and alkyl C-H |
3111-2924 |
|
2 |
C=O Stretching |
Ester and amide carbonyls |
1729-1702 |
|
3 |
C=C Aromatic stretching |
Aromatic rings |
1600-1500 |
|
4 |
NO2 Stretching |
Nitro group |
1350-1300 |
|
5 |
C-O Stretching |
Ester or ether |
1250-1000 |

Graph No.5.2.6: IR Spectrum of 4a 5 (5 -chloro-2-formylbenzamido)-1,3,4-oxadiazolidin-2-yl-4- chlorobenzoate
Table. No.5.2.6: IR Spectrum of 4a 5 (5 -chloro-2-formylbenzamido)-1,3,4-oxadiazolidin-2-yl-4- chlorobenzoate.
|
Sr. No. |
Assignment |
Functional groups |
Wavenumber (cm?¹) |
|
1 |
N–H or O–H stretch |
amide N–H stretch or phenolic O–H |
3300–3400 |
|
2 |
C–H stretch |
Alkyl or aromatic C–H |
2900 |
|
3 |
C=O stretch |
Ester or amide carbonyl group |
1700–1730 |
|
4 |
C=C or C=N stretch |
Aromatic ring or oxadiazole C=N |
1600–1620 |
|
5 |
C–O stretch |
Ester |
1200–1300 |
|
6 |
C–Cl stretch or aromatic C–H bending |
Chlorine-substituted aromatic ring |
700–800 |
NMR Spectrum of 3a 5-(2 -formyl-4-nitrobenzamido)-1,3,4-oxadiaazolidin-2-yl-4-nitrobenzoate
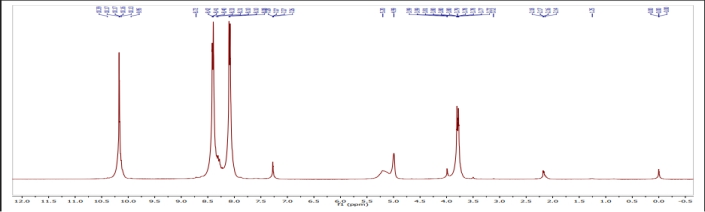
Graph no 5.2.11: NMR Spectrum of compound 3a
Table no. 5.2.11: NMR Spectrum value of compound 3a
|
Sr. No. |
Chemical Shift (δ) |
Assignment |
|
1 |
10.39 |
Aldehyde (-CHO) proton |
|
2 |
10.17 |
Amide NH proton |
|
3 |
9.72 |
Aromatic H ortho to NO? group |
|
4 |
8.46 |
Aromatic protons on substituted benzene rings |
|
5 |
4.99 |
CH/CH? attached to oxadiazole or ester group |
|
6 |
2.18 |
Minor aliphatic protons |
NMR Spectrum of 4a 5 (5 -chloro-2-formylbenzamido)-1,3,4-oxadiazolidin-2-yl-4- chlorobenzoate
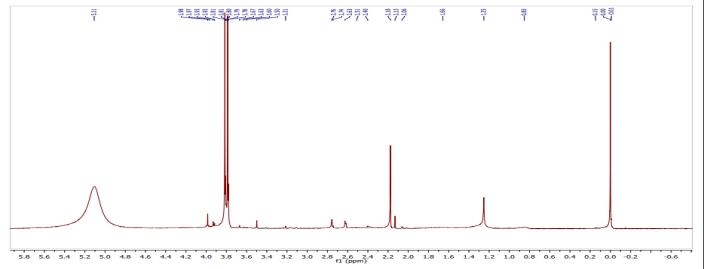
Graph no 5.2.12: NMR Spectrum of compound 4a
Table no.5.2.12: NMR Spectrum value of compound 4a
|
Sr. No. |
Chemical Shift (δ) |
Assignment |
|
1 |
10.38 |
Aldehyde proton (-CHO) |
|
2 |
10.16 |
Amide NH |
|
3 |
8.4 |
Aromatic protons |
|
4 |
5.11 |
amide NH |
|
5 |
3.90 |
CH near oxadiazole or solvent |
|
6 |
2.76 |
trace impurities |
|
7 |
1.25 |
aliphatic solvent |
MASS Spectrum of 3a 5-(2 -formyl-4-nitrobenzamido)-1,3,4-oxadiaazolidin-2-yl-4-nitrobenzoate
Graph no. 5.2.13: MASS Spectrum of compound 3a
MASS Spectrum of 4a 5 (5 -chloro-2-formylbenzamido)-1,3,4-oxadiazolidin-2-yl-4- chlorobenzoate
Graph no 5.2.14: MASS Spectrum of compound 4a
Antimicrobial Activity
Observations:

Fig no.7.7.1: Antimicrobial properties of synthesized compound
Table.no. 7.7.1 Antibacterial screening of synthesized compound (3a-4e) measuring the zone of inhibition in millimeters.
|
Sr.No. |
Compound code |
Zone of inhibition in mm |
|||||
|
E. Coli |
P.aeruginosa |
S.aureus |
|||||
|
100µg/ml |
200µg/ml |
100µg/ml |
200µg/ml |
100µg/ml |
200µg/ml |
||
|
1 |
3a |
10 |
13 |
9 |
15 |
13 |
17 |
|
2 |
3b |
7 |
10 |
5 |
7 |
7 |
9 |
|
3 |
3c |
5 |
9 |
6 |
9 |
7 |
12 |
|
4 |
3d |
4 |
7 |
5 |
7 |
10 |
15 |
|
5 |
3e |
6 |
10 |
5 |
10 |
6 |
9 |
|
6 |
4a |
11 |
14 |
9 |
11 |
12 |
16 |
|
7 |
4b |
7 |
8 |
8 |
7 |
6 |
11 |
|
8 |
4c |
5 |
8 |
6 |
7 |
5 |
7 |
|
9 |
4d |
6 |
9 |
6 |
8 |
4 |
7 |
|
10 |
4e |
5 |
8 |
4 |
9 |
7 |
9 |
|
11 |
Std (Ciprofloxacin) |
12 |
15 |
10 |
13 |
14 |
18 |
Table.no. 7.7.2 Antifungal screening of synthesized compound (3a-4e) measuring the zone of inhibition in millimeters.
|
Sr. No. |
Compound code |
Zone of inhibition in mm |
|||
|
C. albicans |
A. niger |
||||
|
100µg/ml |
200µg/ml |
100µg/ml |
200µg/ml |
||
|
1 |
3a |
14 |
16 |
17 |
20 |
|
2 |
3b |
7 |
8 |
11 |
12 |
|
3 |
3c |
10 |
9 |
9 |
7 |
|
4 |
3d |
12 |
13 |
15 |
10 |
|
5 |
3e |
6 |
6 |
8 |
9 |
|
6 |
4a |
13 |
16 |
18 |
20 |
|
7 |
4b |
5 |
7 |
8 |
9 |
|
8 |
4c |
7 |
8 |
7 |
10 |
|
9 |
4d |
5 |
6 |
8 |
7 |
|
10 |
4e |
6 |
7 |
9 |
12 |
|
11 |
Std (Fluconazole) |
15 |
17 |
20 |
22 |
RESULT AND DISCUSSION
Antimicrobial action: The cup-and-plate method was used to investigate the antibacterial activity of a synthesised chemical. It has been demonstrated that the test sample (3a–4e) shows significant action and a comparable percentage of inhibition to the reference drug.
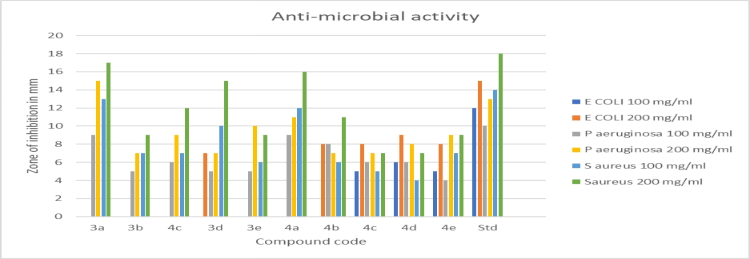
Graph no. 8.3.1 Antibacterial activity on synthesized compound. (3a-4e).
Antifungal activity: To test the synthesized compound's antifungal qualities, the cup-plate method was employed. The test sample (3a–4e) has been seen to have strong activity and a comparable proportion of inhibition to the conventional medication.
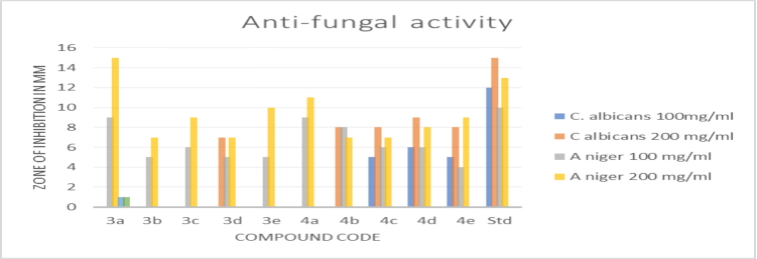
Graph no. 8.3.2 Antifungal activity on synthesized compound. (3a-4e)
SUMMARY AND CONCLUSION
A set of ten synthesized compounds were subjected to molecular docking studies to evaluate their antibacterial and antifungal potential. Compound 4a showed strong binding affinity and significant activity against the E. coli receptor and E. coli GLMU (PDB ID: 4AA7), particularly when combined with an antibacterial inhibitor. Meanwhile, compound 3a demonstrated notable antifungal activity by targeting the receptor DNA gyrase, which contains five disulfide bridges (PDB ID: 1KZN). In the present study, a series of substituted 1,3,4-oxadiazoles were synthesized using microwave-assisted methods and evaluated for their antibacterial and antifungal activities. Microwave synthesis proved to be highly efficient, offering significantly higher yields in a shorter time compared to conventional methods. The synthesized compounds were recrystallized using various solvents, including ethanol, petroleum ether, n-hexane, methanol, butanol, and acetone, through a slow evaporation technique. Among these solvents, ethanol and acetone produced the most well-defined crystal structures, typically forming needle-like, cross-shaped, and clustered crystals. This is particularly important, as polymorphism can greatly influence the bioavailability of drugs—especially those with poor water solubility. The structural integrity of the synthesized compounds was confirmed by satisfactory results from IR, ¹H NMR, and mass spectroscopic analyses. In this study, a group of new oxadiazole compounds was made and tested using both computer-based methods and laboratory experiments. The in-silico (computer) studies showed that some of the compounds could strongly bind to proteins of harmful microbes, suggesting they might work as drugs. Laboratory tests confirmed that a few of these compounds had good antibacterial and antifungal activity. In comparison to ciprofloxacin, compound 4a exhibits substantial antibacterial activity, and In comparison to Fluconazole, compound 3a exhibits similar antifungal activity. Overall, the results suggest that the synthesized oxadiazole derivatives have the potential to be developed into effective medicines for treating infections.
REFERENCES





Namrata Ranpise*, Dr. Shrinivas Mohite, Dr. Sandeep Kane, Akanksha Gadekar, Poorva Kamble, Synthesis in Silico Estimation and Pharmacological Screening of Some Substituted Oxadiazole Derivatives, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 1-13. https://doi.org/10.5281/zenodo.16669728
 10.5281/zenodo.16669728
10.5281/zenodo.16669728