
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmacology, C.T. University, Ludhiana, Punjab, India
Cuproptosis represents a paradigm shift in cancer cell death biology, offering a new therapeutic avenue to overcome multidrug resistance by satisfying basic metabolic requirements, including mitochondrial oxidative phosphorylation and tricarboxylic acid (TCA) metabolism, which are retained even in clinically relevant cancer settings. In contrast to traditional cell death pathways (apoptosis, autophagy, ferroptosis, pyroptosis) cuproptosis is a copper-dependent process in which the Cu 2+ ions directly interact with lipoylated residues of TCA cycle enzymes (especially DLAT and other lipoamidated proteins), leading to protein aggregation, mitochondrial dysfunction, and p53/caspases/BAX/BAK independent immunogenic cell death. Copper ionophores (elesclomol, disulfiram), copper complexes, and nanoparticle-based delivery systems (copper oxide nanoparticles, polydopamine-conjugated carriers, metal-organic frameworks) are pharmacological agents with strong anti-tumor activity in preclinical models and in early clinical trials in cancer biomarkers of drug-resistance, such as triple-negative breast cancer, platinum-resistant ovarian cancer, and hepatocellular carcinoma. The selection of patient stratification key biomarkers (CTR1, FDX1, DLAT expression, and intratumoral copper content) enables the selection of a particular treatment, and the combination approaches with standard chemotherapy, ferroptosis induction, and immunotherapy with checkpoint inhibitors lead to a further increase in treatment efficacy. Although there is positive preclinical and early clinical data, clinical translation of cuproptosis has been highly challenged by several challenges such as narrow therapeutic ranges of copper toxicity, tumor heterogeneity in cuproptosis sensitivity, and development of adaptive resistance; further biomarker identification, nano formulations, combination regimen systematics and mechanistic understanding of resistance mechanisms are essential priorities to develop cuproptosis-based therapies as a transformative element of precision oncology in the treatment-refractory malignancies.
1.1 Definition and significance of cuproptosis
Cuproptosis is considered a paradigm shift in cancer cell death pathways, a novel type of programmed cell death essentially not related to apoptosis, autophagy, necroptosis, pyroptosis or ferroptosis[1]. Initial description: cuproptosis is a copper-dependent mitochondria-centric cell death mechanism, activated by copper overload in cancer cells [2]. As opposed to conventional apoptotic pathways, which require caspases to be activated, cuproptosis is a process where the copper is directly bound to lipoylated residues of the tricarboxylic acid (TCA) cycle enzymes, leading to aggregation of proteins, depolarization of mitochondrial membrane, and programmed cell death[3].
Cuproptosis was revealed as a result of a broad-scale analysis of proteomes and genomes, and indicated that the accumulation of copper induced a unique type of cell death, unlike any of the previously described apoptotic mechanisms. This finding was of great importance as it not only found that copper is a cofactor in enzymatic reactions but also a powerful cytotoxic agent with a new molecular mechanism of killing cells[4]. Cuproptosis has the advantage of bypassing these multidrug resistance mechanisms that cancer cells have developed to avoid conventional cell death mechanisms.
1.2 Overview of drug-resistant cancers
Drug resistance is one of the most overwhelming issues in contemporary oncology, influencing the treatment efficacy and patient outcomes in practically all common types of cancer[5]. The mechanisms of cancer cell resistance are not only interconnected but also multifaceted: genetic mutations to alter drug metabolism, epigenetic silencing of apoptotic processes, expression of drug efflux transporters (especially ATP-binding cassettes), development of DNA repair mechanisms, and reprogramming of metabolic activities in favour of survival under therapeutic stress[6].
Examples of such exceptionally challenging therapeutic issues include triple-negative breast cancer (TNBC), platinum-resistant ovarian cancer, BRAF mutation-related melanoma, and drug-resistant hepatocellular carcinoma[7]. These are cross-resistant to various classes of drugs, and therefore, sequential monopoly is ineffective. This problem is also complicated by the heterogeneity of tumours, where subpopulations of high stemness cancer stem cells and increased drug efflux capacities remain even in the face of chemotherapy and continue to be a source of tumour recurrence[8].
1.3 Current challenges in cancer therapy.
Modern cancer treatment has many daunting challenges in the form of drug resistance. First, cancer cells are multi-targeted and, therefore, when single molecular pathways are inhibited, there is often an activation of other pathways, which makes single-agent therapy inadequate[9]. Second, the emergence of cross-resistance to structurally different chemotherapeutic agents implies basic changes in cellular physiological processes of drug intake and cell death, but not drug-specific ones. Third, epithelial-mesenchymal transition EMT and metabolic plasticity provide cancer cells with the ability to respond quickly to therapeutic forces by changing their oxidative metabolism, lipid composition, and nutrient requirements[10].
Conventional therapy methods have stalled in their effectiveness against resistant cancers. This shortcoming has encouraged the discussion of other forms of cell death, such as ferroptosis, immunogenic cell death, and cuproptosis. Cuproptosis is a more promising pathway since it addresses some of the most basic metabolic requirements of cancer cells, specifically mitochondrial metabolism and copper homeostasis, which is relatively conserved even in drug-resistant forms.
2. COPPER HOMEOSTASIS AND CANCER BIOLOGY
2.1 Copper Metabolism in Normal and Cancer Cells
Copper is also a cofactor in many enzyme systems, such as cytochrome c oxidase (COX), superoxide dismutase (SOD), lysyl oxidase (LOX), and ferroxidase ferroprotein (FPN)[11]. Normal cells have a tight control of copper homeostasis via a coordinated mechanism of copper uptake, intracellular distribution, and excretion. Homeostasis of copper is caused by copper importer CTR1 (copper transporter 1/SLC31A1), which is localized to the plasma membrane and can recognize reduced Cu +, promoting a rapid accumulation in the cell[12].
After internalization, copper is bound to metallochaperones such as ATOX1 (antioxidant 1 copper chaperone), and COX17, which are responsible in transporting copper to particular subcellular compartments. The copper that is intracellular and destined to undergo mitochondrial oxidative phosphorylation is transported to cytochrome c oxidase via particular chaperone pathways. Copper-carrying P-type ATPases (ATP7A and ATP7B) facilitate copper efflux and intracellular sequestration in cases where copper concentrations are above physiological demands[13]. This advanced system regulates cytosolic copper levels to about 10 -15 M without excessive buildup, which would produce excessive reactive oxygen species (ROS).
The metabolism of copper is dramatically changed in cancer cells with copper dependency and dysregulation of homeostatic processes. There are several types of cancer, such as melanoma, lung cancer, hepatocellular carcinoma and breast cancer, where increased levels of copper have been associated in the tumor tissue, relative to the normal adjacent tissue[14]. This build-up of copper is due to the upregulation of CTR1 expression that allows the increase of copper absorption. At the same time, in some instances, the cancer cells tend to show lower ATP7A and ATP7B expression, or vice versa, and this is alongside the dysregulated copper homeostasis[15]. As soon as diagram number 1
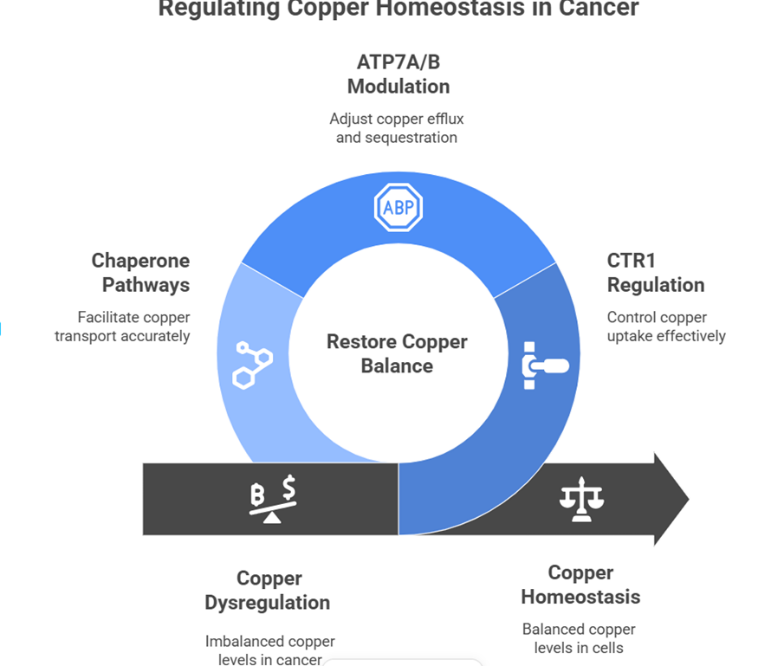
Diagram-1- The diagram illustrates mechanisms that restore copper balance in cancer cells by regulating ATP7A/B, CTR1, and copper chaperone pathways.
2.2 Role of copper dysregulation in cancer development
Copper dysregulation helps to maintain various cancer development and progression hallmarks[16]. High levels of intracellular copper contribute to angiogenesis by activating vascular endothelial growth factor (VEGF) signalling and hypoxia-inducible factor (HIF-1 alpha) and enhancing the neovascularization of tumors [17]. The activation of copper-dependent lysyl oxidase encourages extra-cellular matrix remodeling, which facilitates epithelial-mesenchymal transition and invasion. Also, copper increases the potential of metastasis with the involvement of Wnt/b-catenin signalling and preservation of cancer stem cell populations.
Copper dysregulation, in a mechanistic manner, contributes to cancer cell survival, via various pathways: (1) copper-dependent preservation of mitochondrial respiration that delivers ATP and biosynthetic precursors needed to promote rapid cancer cell proliferation; (2) copper-responsive transcription factors such as hypoxia-inducible factors trigger metabolic reprogramming genes; (3) copper-dependent cross-linking of proteins by lysyl oxidase that maintains tumor microenvironment architecture that facilitates immune evasion[ Ironically, in most situations copper induces cancer formation, but excessive copper buildup over cellular adaptive threshold induces cuproptosis, which is a small therapeutic index of copper-based therapies.
2.3 Copper's dual role in tumor progression and suppression
Copper has been proven to exhibit an outstanding biphasic dose-response profile in cancer biology, where it acts as a promoter and suppressor according to the intracellular concentration[19]. Copper at physiological to moderately elevated concentrations (nanomolar to low micromolar) promotes the survival of cancer cells by causing increased oxidative phosphorylation to enable the metabolic plasticity needed to survive in response to drug treatment and to spread to distant locations. This copper-dependent mitochondrial respiration forms a metabolic dependence the high oxidative phosphorylation-dependent cancer cells are so sensitive to perturbations of copper-dependent mitochondrial complex IV (cytochrome c oxidase).
But at more elevated intracellular levels (near and above 1-2 mM), copper crosses into a survival mode to a potent cytotoxic agent to cause cuproptosis[20]. This change is brought about by copper binding to non-canonical targets, namely lipoylated TCA cycle enzymes such as DLAT (dihydrolipoamide acetyltransferase) and the aggregation of proteins and mitochondrial dysfunction. The therapeutic window that the copper ionophores and copper-based nanotherapeutics use lies between the concentrations of copper that promote survival and those that cause death.
There are adaptive differences in excessive copper in cancer cells. Whilst certain cancer cell lines show robust induction of cuproptosis in response to copper loading, others show relative resistance to cuproptosis by increasing copper efflux (by upregulating ATP7A/B), decreasing copper uptake (by downregulating CTR1), or metabolic reprogramming that decreases the requirement of copper-containing enzyme complexes[21]. These mechanisms of resistance are important to understand the results of clinical responses to copper-based therapies.
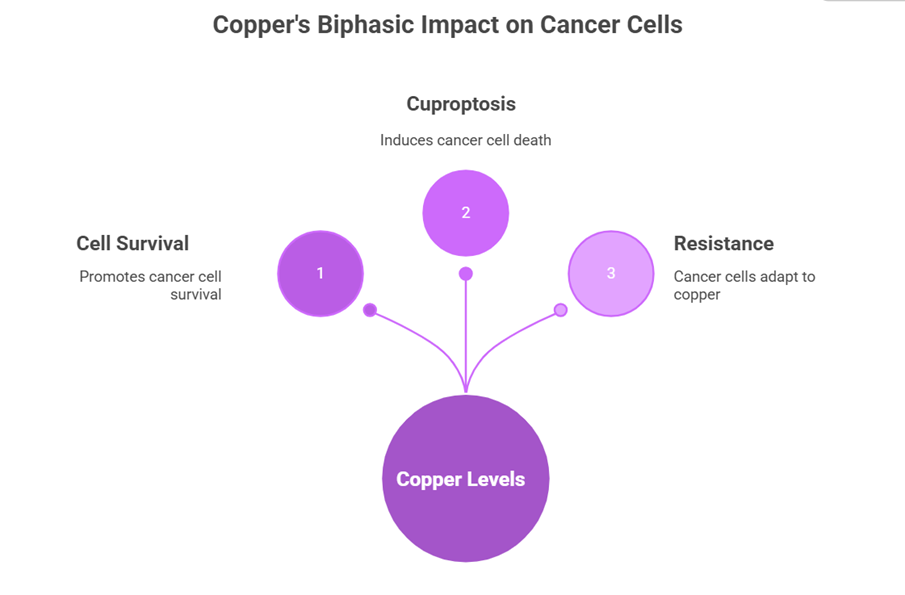
Diagram-2- copper's biphasic effect on cancer cells: low levels promote survival, optimal levels induce resistance adaptation, and high levels trigger cuproptosis death.
3. MOLECULAR MECHANISMS OF CUPROPTOSIS.
3.1 Discovery and definition of cuproptosis (2022 milestone)
The identification of cuproptosis was an important breakthrough in cell death biology. Xu et al. (2022) carried out extensive genomic and proteomic screens that revealed a certain pattern of gene expression and protein changes related to the accumulation of copper that leads to a different type of cell death as compared to apoptosis[22]. Their innovative study proved that cancer cells subjected to copper accumulation showed: (1) maintenance of mitochondrial membrane integrity (not like apoptosis), (2) lack of caspase activity (characteristic of apoptosis), (3) maintenance of autophagy machinery and lack of autophagic flux (characteristic of autophagy-dependent death), and (4) typical patterns of protein aggregation.
Follow-up studies discovered FDX1 (ferredoxin 1) as the key central node that governs the sensitivity to cuproptosis. An increase in FDX1 was associated with increased sensitivity to copper-induced cell death, and FDX1 knockdown was associated with resistance. The finding connected cuproptosis to iron-sulfur cluster homeostasis and mitochondrial redox equilibrium, aiding the formation of the molecular basis of mechanistic knowledge[23].
3.2. Copper binding to lipoylated TCA cycle proteins
Mechanistic Copper binding to lipolylated subunits of TCA cycle enzyme complexes is the cuproptotic centrepiece, which is a new interaction unrelated to the canonical functions of copper as a prosthetic group in metalloenzymes[24]. Proteins that have been lipoylated, such as DLAT (pyruvate dehydrogenase complex component), LIPT1 and LIPT2 (lysophilic acid synthetases), and other lipoylated proteins, have lipoamide cofactors, which are organic dithiol groups and have a high affinity with the Cu2+ ion.
Mechanistically, intracellular copper concentration results in direct binding of Cu2+ ions to the dithiol moiety of lipoamides, resulting in coordinate covalent interactions that alter the conformation and activity of the protein[25]. The result of this copper-lipoamide interaction is resistance of the enzyme catalysis coupled with the resistance of the copper-bound complex to chelation by the endogenous reducing agents. This lipoylated protein selectivity accounts for why cuproptosis targets TCA cycle metabolism, the core energy-yielding machinery of cancer cells, specifically, instead of causing nonselective toxicity. As soon as Figure 3
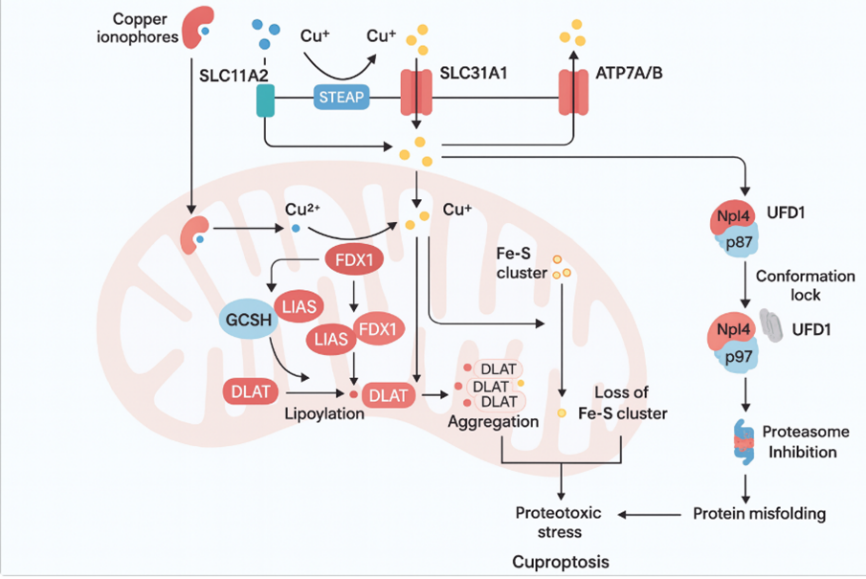
Diagram-3- copper ionophores inducing cuproptosis in cancer cells via Cu transport (SLC31A1), FDX1-mediated lipoylation of DLAT, protein aggregation, and proteostasis collapse.?
3.3 Disulfide bond formation and protein aggregation
Disulfide Bond Formation and Protein Aggregation Disulfide Bond Formation Disulfide bond formation may either be an intraprotein process or an interprotein process. After the copper binds to lipoamide dithiol groups, a cascade of biochemical reactions induces aberrant protein cross-linking and aggregation. The lipoamide complexes are copper-bound and can oxidize, forming intermolecular disulfide bonds between copper-bound proteins and other cellular proteins [26]. This is promoted by a decrease in the antioxidant capacity of cells under the stress of copper, leading to the development of aggregated high-molecular-weight protein molecules that are not degraded by proteases.
These aggregates of proteins are found in specific locations within mitochondria that physically destabilize the ordered quaternary structure needed to allow the normal activity of the enzyme. Proteomic studies also show that lipoylated enzyme complexes clump together with their interacting partners, such as direct interaction with other TCA cycle enzymes and the electron transport chain components[27]. The aggregation process directly disrupts mitochondrial respiration by physically disrupting the architecture of the electron transport chain and TCA cycle enzyme orientation in the same way that protein misfolding stress is produced in neurodegenerative diseases, but on an acute basis in the mitochondrial compartment.
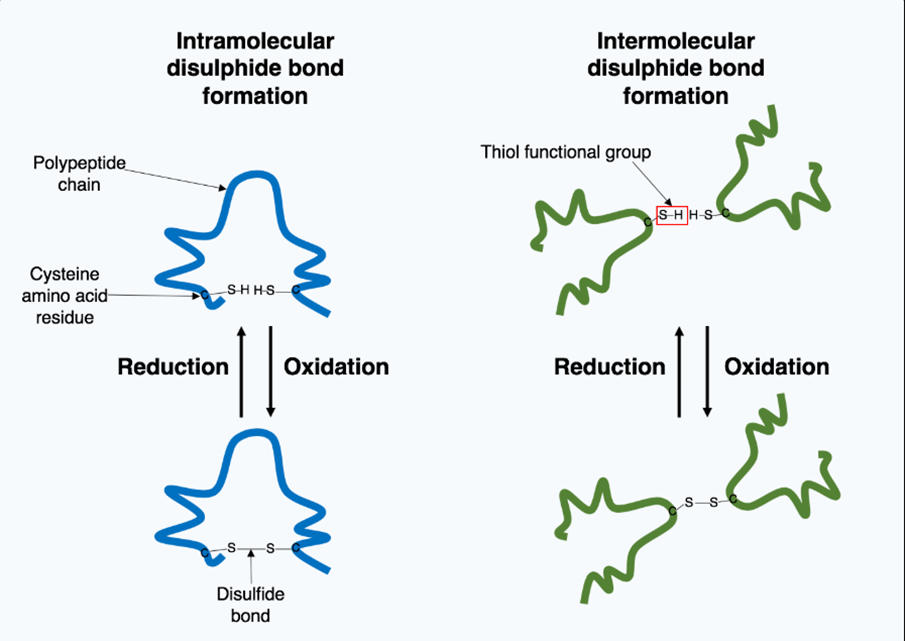
Diagram-4- cysteine thiol oxidation forming intra- and intermolecular disulfide bonds during reduction-oxidation cycles
3.4 Mitochondrial Dysfunction and Cell Death Pathways.
Protein aggregation and destabilization of the lipoylated enzyme complexes cause by copper result in the rapid mitochondrial dysfunction that is characterized by: (1) collapsed mitochondrial membrane potential ( ΔΨm ), (2) descending mitochondrial ATP production, (3) increased mitochondrial ROS generation by partially reduced complexes of the electron transport chain, and (4) release of mitochondrial intermembrane space proteins[28].
Nevertheless, in contrast to apoptosis that occurs via mitochondrial outer membrane permeabilization (MOMP), cuproptosis is also a mitochondrial inner membrane dysfunctional cell death pathway that is not mediated by caspase, nonetheless[29]. The process is through direct interference with the ATP synthase and electron transport chain structure instead of the development of the BAX/BAK-mediated pore. As a result, mitochondrial proteins such as cytochrome c could be released via other means other than via the classical apoptotic pore.
Cuproptotic cells induced by mitochondrial dysfunction downstream activate stress response kinases such as JNK and p38 MAPK, and activate pro-death downstream. The metabolomic results show deep energy deficiency, upstream retention of the TCA cycle compounds in the cases of blocked enzyme activities, and strong redox imbalance. The cells eventually experience cuproptotic death, including loss of cell viability, membrane permeabilization and phagocytic cell clearance patterns that are typical of immunogenic cell death[31].
4. CUPROPTOSIS AND DRUG RESISTANCE
4.1 Relationship Between Copper Metabolism and Chemoresistance
New findings show close links between copper metabolism and the acquisition of treatment resistance[32]. Alterations in copper homeostasis of the drug-resistant cancer cell lines (long-term exposure to chemotherapy) are different in comparison with the drug-sensitive cell lines. In particular, cells of the platinum-resistant ovarian cancer, cisplatin-resistant lung cancer, and 5-fluorouracil-resistant colorectal cancer show low CTR1 expression levels and/or elevated ATP7A/ATP7B expression, which leads to the reduced intracellular copper concentration[33].
This adaptation may indicate that drug-resistant cancer cells that simultaneously acquire drug resistance also reorganize to avoid copper-dependent cell death pathways. The adaptive significance of slower copper uptake could be connected with the lower production of copper-catalyzed ROS in the environment of augmented antioxidant ability, which is typical of a large number of malignant tumors resistant to drugs. Also, ATP7 upregulation in cancer stem cells in heterogeneous tumors results in an increased ability of cells to efflux copper, which is a maintenance mechanism of stemness as well as comparative resistance to induction of cuproptosis[34].
On the other hand, other cancer cell lines with drug resistance retain high levels of CTR1 expression, implying that selective pressure promotes copper uptake as a metabolic benefit to counter the risk of cuproptosis. The heterogeneity justifies the variation in response of cancers of diverse types and tumors of individual patients to copper ionophore therapy.
4.2 Mechanisms of overcoming drug resistance through cuproptosis
The induction of cuproptosis is a new approach towards drug resistance conquest since it alters basic metabolic functions, oxidative phosphorylation by mitochondria and TCA cycle functionality that cannot be lost in drug-resistant cancer cells[35]. Surprisingly, the drug-resistant cancerous cells usually exhibit increased reliance on mitochondrial metabolism relative to their drug-sensitive analogues as a compensatory response to the loss of glycolytic effectiveness. This metabolic reprogramming generates selective susceptibility to the induction of cuproptosis.
There are several ways in which cuproptosis works on drug-resistant cells:
(1) Independence of Distinct Death Mechanism: Cuproptosis starts its activity due to copper-lipoamide interactions that are entirely different to apoptotic, necroptotic, or autophagic. Apoptotic-resistant cancer cells in conventional chemotherapy are still susceptible to cuproptosis since the pathway does not depend on functional p53, intact caspase cascade, or BAX/BAK-mediated mitochondrial outer membrane permeabilization[36].
(2) Mitochondrial Metabolism Dependency: The increased use of drug-resistant cancer cells on mitochondrial oxidative phosphorylation provides a selective vulnerability to copper-mediated mitochondrial complex IV (COX) destruction. This increased metabolic dependence is one Achilles heel, which is an exploitable vulnerability of resistant cancers[37].
(3) Decreased Adaptive Capacity: Although the drug-resistant cells are highly adapted to adapt to the stresses caused by chemotherapy, acute mitochondrial dysfunction as a result of copper-lipoamide binding overwhelms cellular adaptive mechanisms. The high-speed kinetics of inducing cuproptosis, which is resistant to cell activation in 24-48 hours, leave inadequate time to initiate survival processes[38].
(4) Bypassing Efflux Mechanisms: The capacity to induce cuproptosis is still possible in cancer cells with increased drug efflux transporters, even in the absence of drug efflux pump substrates, due to copper ionophores delivery via membrane transport pathways[39].
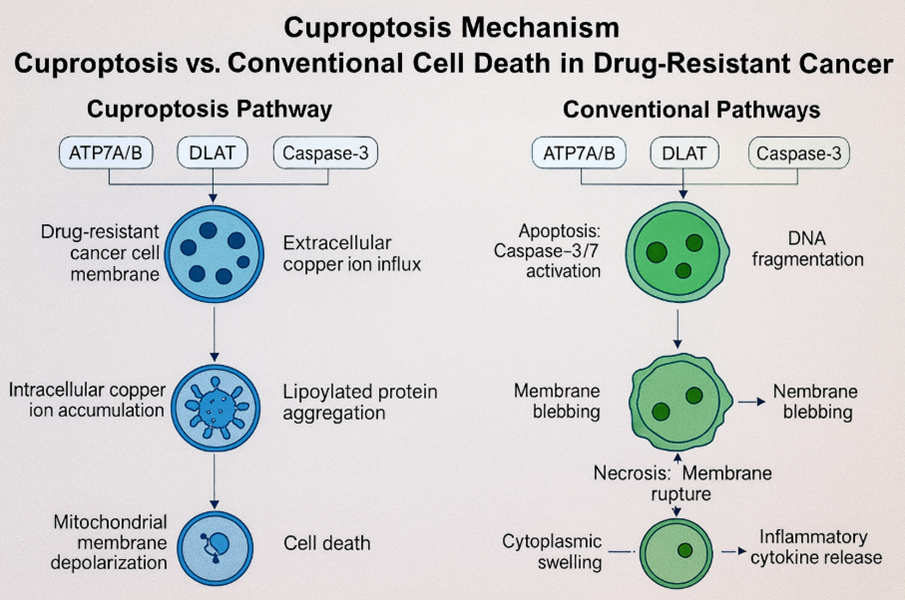
Diagram-5- cuproptosis versus apoptosis and necroptosis in drug-resistant cancer cells, highlighting ATP7B, DLAT, and caspase-3 roles leading to copper-induced cell death.?
4.3 Comparative Analysis of Traditional Cell Death Pathways.
The unique characteristics of Cuproptosis as compared to conventional cell death programs have valuable therapeutic benefits[40]:
Apoptosis: Standard apoptotic pathways in place must involve a functional p53, intact caspase cascades and in certain instances, intact BAX/BAK. Several cancers are drug-resistant, which is caused by apoptosis evasion due to p53 gene mutations, the antiapoptotic overexpression of caspases, inhibition of caspases and the overexpression of antiapoptotic proteins. Cuproptosis does not encounter such evasion.
Autophagy: Although too much autophagy may cause cell death, cancer cells frequently regulate autophagic flux to survive by selecting autophagic cargo. The mitochondrial dysfunction induced by cuproptosis surpasses the ability of cells to autophage mitochondrial aggregates successfully during the window of time in which cell survival is possible[41].
Ferroptosis: Ferroptosis is caused due to iron-dependent lipid peroxidation and inactivation of GPX4. Ferroptosis suppressor protein (FSP1) helps cancer cells to prevent ferroptosis and intensifies antioxidant protection. Direct proteinaceous aggregate formation and mitochondrial complex destruction characterize cuproptosis and avoid iron-dependent oxidative processes[42].
Pyroptosis: The inflammasome activation and gasdermin D pore formation are needed in pyroptosis. Defective inflammasomes have been observed in many cancerous cells and make the cells pyroptosis-resistant. Cuproptosis is independent of the inflammasome signalling [43].
This mechanistic difference is the cause of copper ionophores promoting cell death in cancer cells that were resistant to various conventional and alternative cell death pathways.
|
Cell Death Pathway |
Requirement |
Resistance Mechanism |
Cuproptosis Status |
|
Apoptosis |
p53, Caspases, BAX/BAK |
p53 mutation, Caspase ↓, Bcl-2 ↑ |
? Independent |
|
Autophagy |
Autophagic flux regulation |
Selective cargo handling |
? Overwhelms in 24-48h |
|
Ferroptosis |
GPX4, Iron-dependent ROS |
FSP1 ↑, Antioxidant ↑ |
? Independent |
|
Pyroptosis |
Inflammasome, GSDMD |
Defective inflammasome |
? Independent |
|
Cuproptosis |
Copper, Lipoamides |
None (novel mechanism) |
? Effective |
The Primary Table Source are taken from "Targeting regulated cell death: Apoptosis, necroptosis, pyroptosis, ferroptosis, and cuproptosis", Authors: Z Guo et al.
5. PHARMACOLOGICAL AGENTS FOR INDUCING CUPROPTOSIS
5.1 Copper Ionophores
The most characterized type of cuproptotic agent is copper ionophores. These small molecules are selective in their ability to bind the Cu2+ ions and transport them across the membrane, bypassing the normal copper uptake regulation allowing the rapid and substantial intracellular accumulation, which results in cuproptosis[44].
Elesclomol: Firstly, elesclomol was developed by Synta Pharmaceuticals as a heat shock protein 70 (Hsp70) inducer and ROS generator, but later it was found to be a potent copper ionophore[45]. This ligand-exchange compound is present in a form of equilibrium between the copper-bound state and copper-free form, which allows the idea of copper shuttling across biological membranes. Elesclom is highly selective to cancerous cells compared to normal cells with effective cuproptosis induction at nanomolar to low micromolar concentrations (EC50 values range 50-500 NM, cell type dependent)[46].
Mechanistically, the elesclomol-Cu 2 + complexes bind to CTR1 and promote copper entry into the cytoplasm, where Cu 2 + is reduced to Cu + by ferredoxin reductase (FDX1) and other cytoplasmic reductases[47]. Cu + generated by it has a high affinity to lipoamide dithiol groups, which initiate a cascade of cuproptosis pathways detailed in Section 3. It is important to note that cells that have a high rate of mitochondrial metabolism are particularly sensitive to elesclomol, which is why it is specifically effective against drug-resistant cancers that have high metabolic rates[48]. Elesclomol is safe through clinical trials, but the clinical activity of elesclomol as a mono agent has been shown to be modest in initial clinical trials.
Disulfiram: Disulfiram is an alcohol use disorder therapeutic, FDA-approved copper-chelating compound that acts as a copper ionophore in cancer cells with bioavailable copper[49]. In contrast to elesclomol, disulfiram specifically binds Cu2+ to create lipophilic disulfiram-Cu complexes that easily enter the cell membranes[50]. Upon intracellular dissociation, Cu + releases cuproptosis.
Amazingly, disulfiram-Cu concurrently induces cuproptosis and ferroptosis in hepatocellular carcinoma and other cancer types in different ways: cuproptosis by disrupting the TCA cycle and ferroptosis through iron-dependent lipid peroxidation[51]. This dual cell death killing is the reason why disulfiram is better than elesclomol in various scenarios. Also, disulfiram-Cu beats paclitaxel resistance in triple-negative breast cancer by both triggering cuproptosis and attacking the cancer stem cell via the ALDH+ stem cell inhibition[52].
Other Ionophores: There are other copper ionophores such as thiosemicarbazone derivatives (especially N, N'-bis(2-hydroxybenzylidene) thiosemicarbazide and others), 7-iodo-5-chloro-8-hydroxyquinoline (CQ), pyrithione and other metal-chelating ligands which are ionophores[53]. The compounds tend to share similar principles of mechanism; they are typically copper binding, membrane transport, and intracellular copper liberation, but differ in their strength, selectivity, and toxicity profiles.
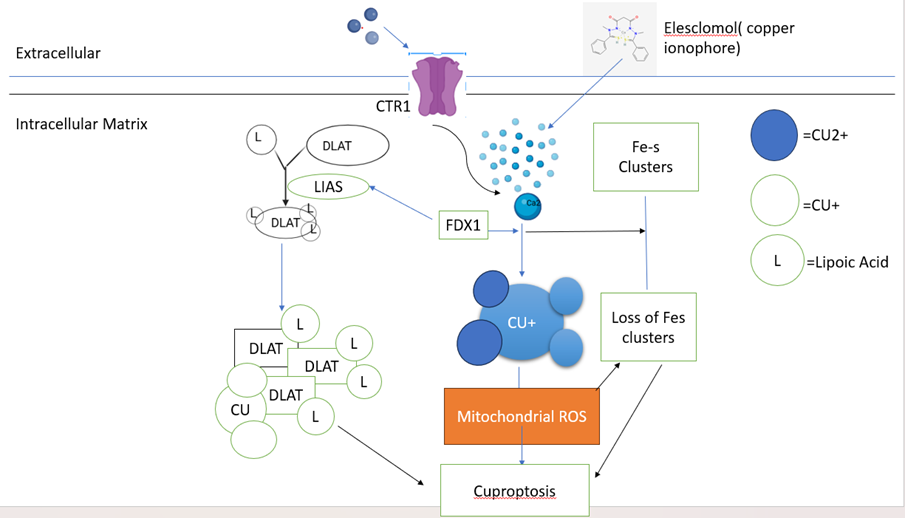
Diagram-6- copper ionophores elevate intracellular Cu via CTR1, FDX1 reduces Fe-S clusters, enabling Cu binding to lipoylated DLAT/LIAS and mitochondrial ROS-induced cell death
5. 2. Copper chelators and their therapeutic applications
Whereas copper ionophores are copper carriers that facilitate intracellular accumulation, copper chelators induce therapeutic effects by the reverse process of sequestration of extracellular and intracellular copper, which inhibits copper-dependent survival signaling[54]. The paradox in therapeutic value of copper chelation under certain situations is associated with the reliance of cancer cells on copper-binding enzymatic activities such as angiogenesis, metastasis, and metabolic plasticity.
Clinically-approved copper chelators such as trientine (triethylenetetramine dihydrochloride) and penicillamine initially used in the management of Wilson disease have a low anticancer effect in preclinical trials. These agents decrease the bioavailable copper, preventing copper-dependent enzymes such as lysyl oxidase and dopamine 2-hydroxylase, inhibiting angiogenesis and metastatic potential[55].
Notably, copper chelation and cuproptosis induction are complementary and not conflicting approaches. Sequential or combinatoric methods, such as pre-treatment of cells with copper which decreases the systemic copper reservoir, and the ensuing administration of copper ionophores with cancer specificity, can maximize therapeutic windows by minimizing the extraneous copper availability in the system whilst still allowing cancer cell-specific copper accumulation enough to induce cuproptosis.
5.3 Copper complexes and dynamic therapy approaches
Combining copper ions with certain organic ligands has been done to generate copper complexes with enhanced selectivity and efficacy[56]. Such engineered complexes tend to include ligands that give:
(1) Targeting moieties Cancer cell-targeting peptides or antibody fragments binding copper complexes will selectively target tumor tissues or cancer cell-specific receptors[57].
(2) Stimulus-responsive properties: PhD-responsive or redox-responsive complexes of copper that release Cu+ in acidic tumor microenvironment, or reduce redox environment, in the intracellular microenvironment[58].
(3) Multifunctional properties: Copper complexes that are conjugated to photosensitizing agents that can be used to provide photodynamic therapy in conjunction with cuproptosis, or copper complexes labeled with radioisotopes that can be used to provide both therapeutic and diagnostic imaging.
One of the most promising methods is photodynamic therapy on the basis of copper ionophore delivery. On light activation, photosensitizers produce singlet oxygen and ROS, which both deplete cellular antioxidant defenses and induce further oxidative stress that combines with mitochondrial dysfunction induced by cuproptosis[59]. These are dynamic therapies, which are the future of copper-based cancer therapy.
6. NANO-BASED DELIVERY SYSTEMS
6.1 Copper-based nanoparticles for targeted delivery
Cuproptosis-inducing agents can now be delivered using nanotechnology to target the tumor selectively using passive and active targeting processes[60]. Copper oxide nanoparticles (CuO NPs) that are directly made of copper have inherent cuproptotic-like activities due to the slow release of copper ions in acidic intracellular vesicles[61]. The accumulation of these particles in the tumors is facilitated by increased permeability and retention (EPR) effect, which then releases the Cu + ions that were deposited in the cancer cells.
Notably, CuO NPs exhibit selective concentration in tumor tissue over normal tissues because of EPR effects and tumor-related elevated vascular permeability[62]. When cancer cells have absorbed acidic lysosomes and phagolysosomes dissolve nanoparticles, the dissolution is catalyzed by acidic lysosomes and phagolysosomes, releasing Cu2+ ions inducing cuproptosis. It has been shown that 22-50 nm CuO NPs would have maximum anti-tumor activity, as larger ones are less effective in tumor penetration and smaller ones have a higher systemic clearance rate prior to tumor retention[63]. As soon as diagram number 7
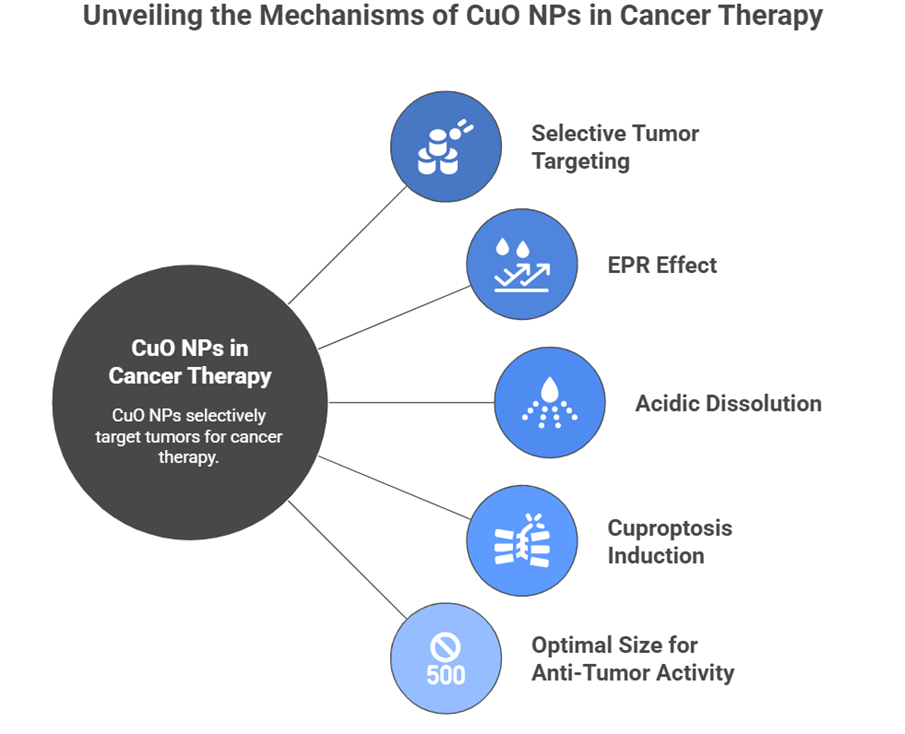
Diagram-7- selective tumor targeting, EPR effect, acidic dissolution, cuproptosis induction, and optimal 500 nm size for anti-tumor activity.?
6.2 Polydopamine and metal-organic frameworks.
Polydopamine (PDA), which is the result of oxidative polymerization of dopamine, is a new biomimetic substance that has outstanding metal-chelating capabilities[64]. The conjugation of PDA nanoparticles with Cu2+ forms Cu2+-polydopamine biocompatible copper delivery systems that have various benefits: (1) polydopamine mussel properties of adhesion enable greater cellular uptake, (2) polydopamine-responsive pH degradation selectively cleanses the acidic tumor microenvironment, releasing Cu2+ intracellularly, (3) polydopamine inherent antioxidant properties cause oxidative stress, enhancing the induction of cuproptosis[65].
Recent works in your research field (polydopamine-based materials) have shown that platelet membrane-coated copper-doped polydopamine nanoparticles (PC NPs) can be used to target tumors exceptionally[66]. The platelet membrane coating gives it immune evasion, which lengthens the circulation of the nanoparticles, and the copper-polydopamine core releases Cu+ in specific acidic pH and high reducing tumor cells. Astonishingly, the single-agent therapy provided by PC NPs minimized the size of the tumor by 1.66-fold in triple-negative breast cancer models without any extra treatment or external activation[67].
Another direction in cuproptosis-inducing nano delivery is based on metal-organic frameworks (MOFs). The crystalline porous structures formed by MOFs that are organic ligands coordinated to copper ions can release copper ions in a controlled manner[68]. MOFs with lipoate analogues or lipoylated peptides exhibit unprecedented selectivity in cuproptosis induction, since only when there are interactions with lipoamides does copper liberation take place. They are materials that allow the tunability of copper release kinetics, by modulation of the structure composition, with an optimization of therapeutic windows[69].
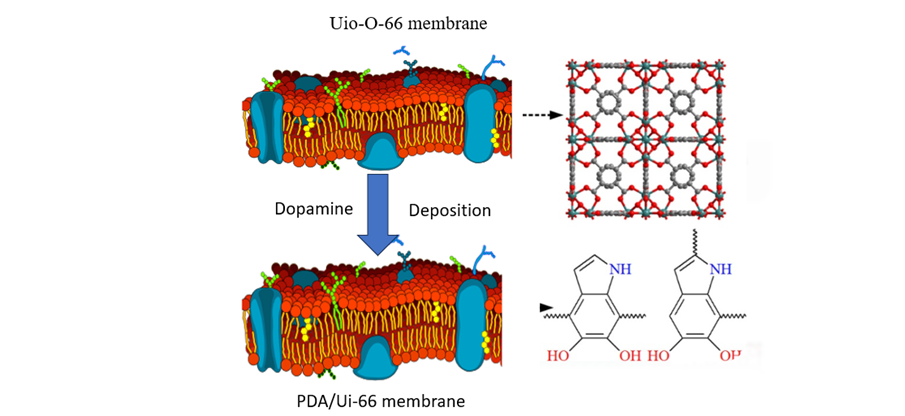
Diagram-8- Dopamine deposition on UIO-66 (red) versus PDA-66 membranes, showing PDA's denser, more uniform coating via self-polymerization
6.3 Enhanced bioavailability and tumor targeting.
Nano-delivery systems significantly increase the bioavailability and tumor selectivity of cuproptosis-inducing agents in several ways[70]:
(1) EPR passive targeting: Nanoparticles 20-200 nm in diameter have a high rate of accumulation in tumors owing to increased vascular permeability and impaired lymphatic drainage with 5-10 fold higher tumor normal tissue ratios than free compounds.
(2) Active targeting Nanoparticles can be functionalized on their surfaces with tumor-targeting ligands (folic acid, transferrin, RGD peptides, antibody fragments) that permit receptor-mediated internalization in cancer cells, increasing selectivity[71].
(3) Membrane coating: Coating of platelet membrane or red blood cell membrane, which provides immune evasion properties, lengthens the circulation half-life of nanoparticles (minutes (naked nanoparticles) versus hours (membrane-coated)) and permits more tumors to accumulate[72].
(4) Stimulus-controlled release: pH-controlled, redox-controlled, or enzyme-controlled nanoparticles release cuproptosis-inducing agents directly on tumor microenvironments, decreasing systemic exposures and off-target toxicity[73].
The advances allow the attainment of effective levels of intracellular copper concentrations which induce cuproptosis and which cause minimal systemic toxicity- an important factor since copper has crucial physiological functions.
7. SYNERGISTIC THERAPEUTIC STRATEGIES.
7.1 Combination of cuproptosis with ferroptosis induction
Increasingly, a combination of Cuproptosis with Ferroptosis Induction is adopted to address issues of plasminogen preservation, confirmed by direct imaging of the cellular nucleus and cytosol, and the precise locations of plasminogen.
Ferroptosis is an iron-dependent cell death that requires the inactivation of GPX4 followed by lipid peroxidation, which may be synergistically induced together with cuproptosis to induce cancer cell death[74]. The mechanisms behind synergy are:
(1) Complementary oxidative stress: ROS generation induced by copper and lipid peroxidation catalyzed by iron increases intracellular oxidative stress that surpasses cellular antioxidants[75].
(2) Glutathione depletion: Both cuproptosis and ferroptosis deplete cellular glutathione (GSH)-cuproptosis by reducing Cu +, and ferroptosis by catalyzing GPX4 reactions-forming a simultaneous severe redox imbalance[76].
(3) synergy of mitochondrial dysfunction: copper caused the impairment of complex IV in synergy with the damage of complex I/III caused by iron, resulting in severe energy alteration[77].
(4) Non-redundant death pathways: Cuproptosis by protein aggregation and ferroptosis by lipid peroxidation are mechanistically different processes that do not allow the pathways to be compensated[78].
This combination has been clinically translated to include disulfiram-Cu with ferroptosis inducers (e.g., imidazole ketone erastin, IKE) to effectively kill drug-resistant cancer cells, such as myelodysplastic syndromes, inaccessible to other methods[79].
7.2 combination with Conventional Chemotherapy.
Combination therapy with cuproptosis-promoting agents and standard chemotherapy shows Synergistic interactions that show a stronger treatment effect by various mechanisms[80]:
Chemosensitization: Autophagy is blocked by copper ionophores such as elesclomol and disulfiram by activating the mTOR pathway through DLAT and decreasing the ability of cancer cells to autophage damaged organelles during chemotherapy. Such chemosensitization effect increases the efficacy of taxanes and platinum agents and other chemotherapies[81].
Stemness Suppression: Disulfiram-Cu is an agent that specifically acts on the cancer stem cell (ALDH+) by weakening aldehyde dehydrogenase and stem cell-related transcription factors, leading to a decrease in the number of stem cells that can normally mediate chemotherapy resistance[82].
Metabolic Reprogramming: Cuproptosis-stimulating agents block glycolysis and activate oxidative phosphorylation, inducing metabolic conditions that increase the effectiveness of chemotherapy against cells that have switched to glycolytic metabolism[83].
G2/M Phase Arrest: Elesclomol-Cu disrupts cell autophagy and facilitates G2/M cell cycle arrest via mTOR signals and hyperirritates cancer cells to cell-cycle phase-dependent chemotherapy, such as taxanes[84].
7.3 Enhanced immunotherapy through metal-dependent cell death
Cuproptosis is a type of immunogenic cell death (ICD), which is initiated by danger-associated molecular patterns (DAMPs), such as ATP, calreticulin, and high-mobility group box 1 (HMGB1)[85]. The generation of anti-tumor immune response by the activation of antigen-presenting cell maturation and cross-presentation is produced by these DAMPs, which supplement the direct elimination of the cancer cells via cuproptosis.
Combination of cuproptosis induction and checkpoint immunotherapy (anti-PD-1/PD-L1) show improved anti-tumor efficacy by a variety of mechanisms[86]:
(1) PD-L1 inhibition: Metabolic re-programming and stress signalling caused by copper decrease PD-L1 levels by modulating the HIF-1 and NF-KB pathways, and increasing the effect of immunotherapy with immune checkpoint blockers[87].
(2) DAMP-regulated DC maturation. Immunogenic cuproptotic cell death produces an inflammatory microenvironment that facilitates the maturation of dendritic cells and Th1-biased anti-tumor immunity[88].
(3) Remodelling of the tumour microenvironment: Cancer cell elimination mediated by cuproptosis eliminates immune-suppressive metabolic factors (lactate, tryptophan metabolites) and suppresses the activation of cancer-associated fibroblasts, forming a permissive immune microenvironment[89].
Notably, disulfiram-Cu plus anti-PD-L1, which is sensitive to PD-L1 therapy in NSCLC regardless of ATP7B, is regulated by the HIF-1 pathway, and provides clinical potential of this combination[90].
8. CANCER TYPE-SPECIFIC APPLICATIONS
|
Cancer Type |
Leading Agent |
Resistance Mechanism |
Key Biomarker |
Clinical Status |
|
Triple-Negative Breast Cancer |
Disulfiram-Cu |
ALDH+ stem cells, ATP7B upregulation |
CTR1, ALDH activity |
Preclinical optimization |
|
Platinum-Resistant Ovarian Cancer |
Elesclomol-Cu |
CTR1 downregulation, ATP7 upregulation |
CTR1, FDX1, ATP7A/B |
Phase II (pending results) |
|
Cisplatin-Resistant Lung Cancer |
Elesclomol |
MITF amplification, metabolic plasticity |
FDX1, DLAT, MITF |
Preclinical + Phase I combination |
|
BRAF-Mutant Melanoma |
Disulfiram-Cu |
Vemurafenib resistance, EMT phenotype |
PD-L1, SNAIL1, CTR1 |
Preclinical studies |
|
Hepatocellular Carcinoma |
Disulfiram-Cu |
HBV/HCV-associated proliferation, Wnt signaling |
HIF-1α, CTR1, ATP7B |
Preclinical + early clinical |
|
Colorectal Cancer (5-FU resistant) |
Elesclomol |
Thymidylate synthase upregulation, YTHDC2 downregulation |
YTHDC2, CDKN2A, CTR1 |
Preclinical studies |
|
Castration-Resistant Prostate Cancer |
Elesclomol-Cu + Enzalutamide |
AR pathway independence, NUDT21-mediated resistance |
NUDT21, FDX1, DLAT |
Phase I/II combinations |
|
Myelodysplastic Syndromes |
Elesclomol-Cu + IKE |
Impaired differentiation, ferroptosis resistance |
DLAT, TP53 mutation status |
Preclinical studies |
Details taken from- Redox Biol (2023) "FDX1-dependent mechanisms of elesclomol-Cu" [Zulkifli Metal.], "Novel insights into elesclomol anticancer mechanisms" [Gao J et al.], Elesclomol induces copper-dependent ferroptosis/ cuproptosis" [Gao W et al.] "Advancing Cancer Therapy with Copper/DSF" [Skrott et al.]
8.1 Breast cancer and triple-negative breast cancer
Triple-negative breast cancer (TNBC) is the worst prognosis and the most aggressive type of breast cancer that has a high likelihood of resistance to chemotherapy [91]. The inherent chemoresistance of TNBC indicates a number of factors: impaired p53 activity, the activation of the PI3K pathway that facilitates survival cues, high tumor heterogeneity with large cancer stem cell groups, and frequent down-regulation of CTR1[92].
Disulfiram-Cu therapy proves to be extremely effective against TNBC in a variety of mechanisms[93]. To begin with, disulfiram-Cu has cuproptotic activity by way of copper ionophore. Second, disulfiram, by itself, is an inhibitor of an enzyme, aldehyde dehydrogenase (ALDH), preferentially expressed in ALDH+ cancer stem cells, which mediate TNBC chemotherapy resistance. Third, disulfiram-Cu reconfigures the vulnerability of ALDH+ stem cells to cisplatin, allowing re-sensitisation of platinum-resistant cells[94].
Preclinical analyses prove that disulfiram-Cu, as a single agent, shrinks the tumor of TNBC by 50-70% in xenograft models. It can be combined with paclitaxel to increase efficacy, which overcomes paclitaxel resistance with the induction of cuproptosis and suppression of ALDH+ stem cells. Multi-centre trials are being conducted to translate clinical trials[95].
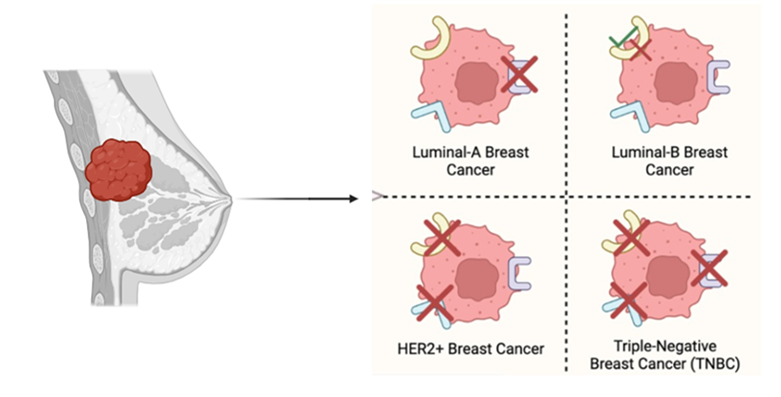
Diagram-9- Breast cancer subtypes from luminal tumor: LumA and LumB (HER2-) cancers, HER2+ breast cancer, and triple-negative breast cancer (TNBC).?
8.2 Lung cancer, melanoma, and hepatocellular carcinoma
Lung Cancer: FDX1 is highly expressed in cisplatin-resistant squamous cell carcinoma of the lung, especially those containing EGFR mutations or ALK rearrangements, which are resistant to tyrosine kinase inhibitors, and are sensitive to elesclomol-Cu[96]. Elesclomol-Cu triggers a strong cuproptosis in both NSCLC and SCLC models with a high level of effectiveness on cells that have a high level of mitochondrial metabolism (which is commonly typical of drug-resistant variants). Phase I combination trials of elesclomol and standard chemotherapy are being carried out.
Melanoma: Resistance to Vemurafenib harbouring BRAF mutation melanoma with upregulation of disulfiram-Cu by dual ferroptosis and cuproptosis elevation, as well as suppression of EMT-linked transcriptional factors upholding resistance[97]. Disulfiram-Cu is undergoing clinical studies in melanoma; preliminary trials have shown melanoma disease to be stabilized in patients who were developing at a rapid rate before.
Hepatocellular Carcinoma: Hepatocellular carcinoma (HCC) is one of the most promising indications in the induction of cuproptosis because: (1) Hepatocellular carcinoma (HCC) tissue has a high baseline level of copper, (2) HCC cells almost inevitably rely on mitochondrial oxidative phosphorylation, (3) HBV/HCV-related tumorigenesis imposes certain metabolic requirements that cuproptosis can be used to satisfy[98]. At the same time, disulfiram-Cu triggers cuproptosis and ferroptosis in HCC models that show 70-80% tumor growth inhibition in xenografts[99]. Preclinical trials in high-stage HCC are encouraging.
Diagram-10- primary lung tumor metastasizing to liver via circulation, with associated cells like tumor cells, macrophages, Kupffer cells, and stellate cells
8.3 Colorectal cancer and other malignancies.
Colorectal cancer that is resistant to 5-fluorouracil has the altered expression of CDKN2A and YTHDC2, which are related to the increasing sensitivity to elesclomol-Cu[100]. The adaptation mechanisms, which include the upregulation of thymidylate synthase, convert cells into glycolysis-independent and oxidative phosphorylation-dependent cells, which poses a metabolic weakness to the induction of cuproptosis[101].
Other malignancies that are sensitive to cuproptosis include castration resistant prostate cancer (CRPC), in which the combination of elesclomol-Cu and ferroptosis induction (IKE) induces effective differentiation and apoptosis of differentiation-resistant tumour cells[102], and myelodysplastic syndromes, in which the combination of elesclomol-Cu and ferroptosis induction (IKE) results in potent differentiation and apoptosis of differentiation-resistant tumor cells[103].
9. THE BIOMARKERS AND DETECTION METHODS.
9.1 Cuprotic Induction Measuring Techniques.
Strong evaluation tools to measure cuproptosis induction are needed to develop preclinical and clinical translation. Current techniques include:
Flow cytometry Cuproptotic cells have typical phenotypes such as loss of mitochondrial membrane potential (measured by TMRM or TMRE staining), lack of Annexin V binding (not apoptosis), and alteration of membrane permeability measured by propidium iodide uptake[104].
Transmission Electron Microscopy (TEM): Ultrastructural examination demonstrates cuproptosis-related morphology, which includes a disruption of mitochondrial cristae, accumulation of protein aggregates in mitochondria, and retained nuclear architecture[105].
Immunofluorescence Microscopy: Cellular-resolution measurement of cuproptosis induction can be achieved by direct visualization of protein aggregates using ubiquitin staining, lipoylated protein staining, and aggregation-specific antibodies[106].
Metabolomics and Proteomics: Metabolic signatures such as TCA cycle impairment, pyruvate build-up, lactate, and citrate are observed by LC-MS/MS analysis with proteomic evidence of lipoylated protein aggregation[107].
Real-Time Cellular Analysis: Impedance-based biosensors that allow monitoring cell death kinetics caused by cuproptosis in real-time, which is better than end-point viability assays to determine temporal dynamics[108].
9.2 Cuproptosis-related gene identification
Extensive transcriptomic and genomic studies have defined the major genes that are involved in the maintenance of cuproptosis sensitivity[109]:
Copper Homeostasis Genes: CTR1 (copper uptake), ATP7A/B (copper efflux), ATOX1(copper trafficking) where CTR1 is downregulated (linked with cuproptosis resistance)[110].
Cuproptosis Core Genes: The FDX1 (ferredoxin 1) upregulation is associated with cuproptosis sensitivity, and the upregulation of DLAT (dihydrolipoamide acetyltransferase) increases cuproptosis sensitivity[111].
Metabolic Genes: CDKN2A, which controls glycolysis and copper homeostasis, genes that control dependence on oxidative phosphorylation[112].
Regulatory Genes: FOXO3 transcription factor, whose high levels in the nucleus inhibit FDX1 and protect against cuproptosis, NUDT21 that facilitates enzalutamide and cuproptosis resistance in prostate cancer[113].
Immune Regulation: CEBPB and inflammatory response genes that regulate the immunogenicity of cuproptosis[114].
Gene signature analysis makes it possible to predict personalized sensitivity to cuproptosis of patients/tumors and therefore choose a personalized therapy.
|
Category |
Techniques/Markers |
Description |
Application / Significance |
|
Cuproptosis Measuring Techniques |
Flow Cytometry |
Loss of mitochondrial membrane potential (TMRM/TMRE staining), lack of Annexin V binding, propidium iodide uptake |
Identifies distinct cell death phenotype of cuproptosis |
|
|
Transmission Electron Microscopy (TEM) |
Disruption of mitochondrial cristae, protein aggregate accumulation, normal nuclear structure |
Morphological confirmation of cuproptosis |
|
|
Immunofluorescence Microscopy |
Visualization of protein aggregates using ubiquitin, lipoylated proteins, specific antibodies |
Cellular-level detection of cuproptosis-related proteins |
|
|
Metabolomics and Proteomics |
TCA cycle impairment, pyruvate buildup, lactate, citrate analysis by LC-MS/MS, lipoylated protein aggregation |
Detects metabolic and proteomic changes associated with cuproptosis |
|
|
Real-Time Cellular Analysis |
Impedance-based biosensors for monitoring cell death kinetics |
Dynamic monitoring of cuproptosis over time |
|
Cuproptosis-Related Genes |
Copper Homeostasis Genes (CTR1, ATP7A/B, ATOX1) |
CTR1 downregulation linked with cuproptosis resistance |
Copper uptake and trafficking |
|
|
Core Genes (FDX1, DLAT) |
Upregulation increases cuproptosis sensitivity |
Essential for cuproptosis induction |
|
|
Metabolic Genes (CDKN2A) |
Controls glycolysis and copper homeostasis |
Metabolic regulation linked to cuproptosis |
|
|
Regulatory Genes (FOXO3, NUDT21) |
FOXO3 inhibits cuproptosis; NUDT21 linked to resistance |
Regulate sensitivity and resistance to cuproptosis |
|
|
Immune Regulation Genes (CEBPB, inflammatory genes) |
Control immunogenicity of cuproptosis |
Affect immune response in tumors |
|
Clinical Biomarker Development |
CTR1 Expression |
Tumor CTR1 expression correlates with elesclomol and disulfiram response |
Patient stratification and therapy sensitivity prediction |
|
|
FDX1 Expression |
Predictor of cuproptosis sensitivity across cancer types |
General biomarker of cuproptotic susceptibility |
|
|
Copper Content |
Measured by X-ray fluorescence microscopy or ICP-MS |
Identifies tumors prone to cuproptosis |
|
|
Metabolic Biomarkers |
FDG-PET and lactate imaging detect tumors reliant on glycolysis or oxidative phosphorylation |
Predictors of cuproptosis sensitivity |
|
|
Gene Expression Signatures |
Composite signatures using CTR1, FDX1, DLAT, FOXO3, others |
Personalized prediction of tumor sensitivity to cuproptosis |
Details taken from- "Recent progress of methods for cuproptosis detection" [Li et al.] ", Cuproptosis: molecular mechanisms and therapeutic landscape" [Wang et al.] ", Cuproptosis: molecular mechanisms, cancer prognosis" [Zhang et al.] ? ", Decoding cuproptosis and cuproplasia" [Liu et al.]
9.3 Clinical biomarker development
Cuproptosis research needs to be translated to clinical application by creating strong biomarkers to predict response to therapy:
CTR1 Expression: The expression of tumor CTR1 in immunohistochemistry is associated with the response of elesclomol and disulfiram, and thus, patient stratification is possible. CTR1-high tumors are not only sensitive to cuproptosis, but CTR1-low tumors are also relatively resistant[115] to cuproptosis.
FDX1 Expression: Tumor FDX1 expression is a predictor of cuproptosis sensitivity not dependent on the type of cancer, and possibly a general cuproptotic sensitivity biomarker[116].
Copper Content: Direct determination of intratumoral copper by X-ray fluorescence microscopy or inductively coupled plasma mass spectrometry (ICP-MS) of microscopic analyses of tumor samples with copper-containing basal copper determines tumors that are prone to the induction of cuproptosis[117].
Metabolic Biomarkers: Positron emission tomography using fluorodeoxyglucose (FDG-PET) and lactate imaging detect tumors that rely on glycolysis or oxidative phosphorylation, respectively, as predictors of cuproptosis sensitivity, because tumors that rely on oxidative phosphorylation are the most susceptible to cuproptosis[118].
Gene Expression Signatures: Signatures constructed of multiple genes, using CTR1, FDX1, DLAT, FOXO3, and other regulatory genes, allow individual tumor cuproptosis sensitivity to be predicted in an advanced way[119].
10. CLINICAL TRANSLATION AND FUTURE PROJECTIONS.
10.1 Current clinical investigations and trials
Cuproptosis-inducing agents are being tested in multiple clinical trials, but there is little published evidence as the field has only recently developed[120]:
Elesclomol Clinical Program: Phase I clinical trials established elesclomol safety up to 8-10 mg/kg, with its major adverse effects being peripheral neuropathy (reversible) and infrequent instances of hepatotoxicity[121]. A phase II trial of elesclomol, combined with paclitaxel in platinum-resistant ovarian cancer (NCT identifier under investigation) demonstrated potentially promising but modest clinical benefit with disease stabilization in about 30% of platinum-resistant patients[122].
Disulfiram Re-purposing: Disulfiram with copper supplementation has been studied in several Phase I/II trials in various cancer types such as TNBC, NSCLC and HCC. Preliminary experiences indicate that there is tolerability that is acceptable, and certain cases of objective tumor response[123].
Combination Approaches: Phase I trials of elesclomol with immunotherapy (e.g., anti-PD-L1), with conventional chemotherapy, with other agents are currently in the process of starting enrollment. These are preliminary studies to determine safety and initial efficacy preceding the Phase II study[124].
10.2 Challenges in translating research to practice
Although the preclinical and early clinical evidence is encouraging, there are several obstacles to clinical translation[125]:
Bioavailability and Pharmacokinetics Elesclomol and other copper ionophores have unstable bioavailability and pharmacokinetics based on food consumption, GI pH, and interpersonal differences. Optimization of formulations that allow constant systemic exposure is still in progress[126].
Copper Toxicity: Systemic copper toxicity is a severe limitation because an adequate concentration of copper intracellularly to induce cuproptosis may lead to accumulation of copper in body organs such as the liver and nervous system[127]. This extremely small therapeutic index requires advanced delivery techniques that allow selective accumulation of copper in cancer.
Heterogeneous Response to Cuproptosis: The responses to cuproptosis differ in single tumors across the expression of CTR1, levels of FDX1, baseline quantities of copper content, and metabolic condition. Predictive biomarkers that can be used to stratify patients are still being developed[128].
Resistance Evolution: The long-term efficacy of the resistance to cuproptosis-inducing agents is potentially restricted due to the selection of cancer cells that become more efficient in copper efflux or less reliant on oxidative phosphorylation[129].
Drug-Drug Interactions: The interactions of copper ionophores with chemotherapy drugs, checkpoint inhibitors, and other therapeutics should be systematically studied to maximize the combination regimens[130].
10.3 Future Directions and Research Priorities.
The areas of future research need to include[131]:
(1) Biomarker Discovery: A global description of biomarkers that predict cuproptosis sensitivity, making it possible to stratify the patient and tailor therapy[132].
(2) Nanoformulation Optimization: Design of superior nanoparticles with improved tumor targeting, stimuli-responsive copper release and favourable pharmacokinetics[133].
(3) Optimization of Combinations: Systematic assessment of cuproptosis induction with chemotherapy, immunotherapy, targeted therapy, and so on to find the best therapeutic regimes and dosages[134].
(4) Resistance Mechanism Elucidation: Resistance mechanisms to cuproptosis, and devising a plan to overcome resistance evolution[135].
(5) Mechanistic Refinement: More detailed description of cuproptosis molecular mechanisms, especially how different types of cancer respond differently to it, and what are targetable vulnerability factors[136].
(6) Clinical Trial Expansion: Phase II/III trials that characterize the actual clinical efficacy of cuproptosis-inducing agents in each cancer type, paying close attention to patient selection with respect to new biomarkers[137].
(7) Immunogenic Cell Death Optimization: The maximization of immunogenicity of cell death induced by cuproptosis by optimal use of combinations with checkpoint immunotherapy and cellular immunotherapy strategies[138].
CONCLUSION
Cuproptosis is a paradigm shift in cancer cell death biology, which provides new therapeutic approaches to overcome multidrug resistance by controlling the basic metabolic processes, namely, mitochondrial oxidative phosphorylation and TCA cycle activity, which are still required, even in cancer cells resistant to chemotherapy. Mechanistic uniqueness of cuproptosis with respect to conventional apoptotic, autophagic and ferroptotic pathways means that cell lines resistant to conventional cell death mechanisms are susceptible to induction of cuproptosis.
Copper ionophores (elesclomol, disulfiram, thiosemicarbazones), copper complexes and novel nanoparticle-based delivery vehicles are shown to be exceptionally effective in preclinical models and in initial clinical studies. Of particular interest are the synergistic interactions of cuproptosis induction with other forms of chemotherapy, ferroptosis induction, and checkpoint immunotherapy and optimism that therapeutic resistance can be surmounted by using several distinct simultaneous modes of cell death and immune activation.
Nevertheless, clinical translation has significant issues, such as the existence of narrow therapeutic indices of copper dosing, the need to predict tumor responses with predictive biomarkers, and the evolution of resistance necessitating long-term mechanistic insights. Additional research agendas involve the identification of biomarkers that can be used to stratify patients, optimization of nanoformulations that can be used to selectively deliver copper to cancer, a thorough assessment of combination therapies, and a systematic study of cuproptosis-resilient variants.
With the accumulation of clinical experience and the identification of biomarkers that predict cuproptotic sensitivity, cuproptotic-based therapies will become significant elements of personalized cancer treatment therapies, especially in drug-resistant malignancies, as the most difficult treatment issues in modern oncology.
REFERENCES





Bijoy Ghosh, Meckie Khaliz Chapola, Anurag Biswas, Saurav Kumar, Nancy, Hildegatha Mgaya, The Pharmacological Induction of Cuproptosis: A Novel Strategy for Targeting Drug-Resistant Cancers, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 2695-2723. https://doi.org/10.5281/zenodo.18360645
 10.5281/zenodo.18360645
10.5281/zenodo.18360645