
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
JGVVSS'S Suyash College Of Pharmacy, Warud B. K Jalna 431206,Maharastra
This review determines the various stages of drug discovery, pre-clinical trials and clinical trials. This process involves the identification of chemical compound, synthesis, characterization, validation, optimization, screening and assay for therapeutic efficacy. Once a new drug target or promising molecule has been identified the process of moving from the science laboratory to the pre-clinical to clinical trials have been discussed in this article. The main aim of this process is identifying the chemical compound which is therapeutically useful in treating and curing the disease. The most common steps in the development of a new drug are discovery or synthesis of a potential new drug compound or elucidation of a new drug. Once a chemical compound has shown its therapeutic efficacy in these investigations, it will initiate the process of drug development earlier to clinical trials. The drug development from initial idea to the market is a very complex process which can take upto 5 to 10 years and cost of $17 billion. Due to high budget of research & development and clinical trials drug discovery process is the most expensive. The average time taken for the drug discovery is almost 12 - 15 years to develop a single new compound and enter into market.
Drug discovery has a long history and dates back to the early days of human civilization. In those ancient times, treatments were often discovered by chance or resulted from observation of nature, typically but not exclusively, using ingredients extracted from plants/animals, and not just used for physical remedy but also for spiritual healing. Modern drug discovery research started to being performed around the early 1900s.
Drug discovery is a multifaceted process, which involves identification of a drug chemical therapeutically useful in treating and management of a disease condition. Typically, researchers find out new drugs through new visions into a disease process that permit investigator to design a medicine to stopover or contrary the effects of the disease. The process of drug discovery includes the identification of drug candidates, synthesis, characterization, screening, and assays for therapeutic efficacy. When a molecule avails its
satisfactory results in these investigations, it will commence the process of drug development subsequent to clinical trials. Drug discovery and development is an expensive process due to the high budgets of R&D and clinical trials. It takes almost 12-15 years to develop a single new drug molecule from the time it is discovered when it is available in market for treating patients. The average cost for research and development for each efficacious drug is likely to be $900 million to $2 billion. This figure includes the cost of the thousands of failures: For every 5,000-10,000 compounds that enter the investigation and development pipeline, ultimately only one attains approval. These statistics challenge imagination, but a brief understanding of the R&D process can explain why so many compounds don‘t make it and why it takes such a large, lengthy effort to get one medicine to patients. The Success requires immense resources the best scientific and logical minds, highly sophisticated laboratory and technology; and multifaceted project management. It also takes persistence and good fortune. Eventually, the process of drug discovery brings hope, faith and relief to billions of patients.
Stages of drug discovery and development include:

Figure 1: Stages of drug development and timeline
Target Identification
The first step in the discovery of a drug is identification of the biological origin of a disease, and the potential targets for intervention. Target identification starts with isolating the function of a possible therapeutic target (gene/nucleic acid/protein) and its role in the disease. Identification of the target is followed by characterization of the molecular mechanisms addressed by the target. An ideal target should be efficacious, safe, meet clinical and commercial requirements and be druggable. The techniques used for target identification may be based on principles of molecular biology, biochemistry, genetics, biophysics, or other disciplines.
Approaches:
Target Validation
Target validation is the process by which the expected molecular target – for example gene, protein or nucleic acid of a small molecule is certified. Target validation includes: determining the structure activity relationship (SAR) of analogs of the small molecule; generating a drug-resistant mutant of the presumed target; knockdown or over expression of the presumed target; and monitoring the known signalling systems downstream of the presumed target. Target validation is the process of demonstrating the functional role of the identified target in the disease phenotype. Whilst the validation of a drug‘s efficacy and toxicity in numerous disease-relevant cell models and animal models is extremely valuable – the ultimate test is whether the drug works in a clinical setting.
Target validation can be broken down in to two key steps.
Reproducibility:
Once a drug target is identified, whether it be via a specific technique or from review of literature, the first step is to repeat the experiment to confirm that it can be successfully reproduced. The target validation technique includes affinity chromatography, expression-cloning, protein microarray, reverse transfected cell microarray, biochemical suppression, siRNA, DNA microarray, system biology and study of existing drugs. Introduce variation to the ligand (drug)-target environment
Identification of Lead A chemical lead is defined as a synthetically stable, feasible, and drug like molecule active in primary and secondary assays with acceptable specificity, affinity and selectivity for the target receptor. This requires definition of the structure activity relationship as well as determination of synthetic feasibility and preliminary evidence of in vivo efficacy and target engagement. Characteristics of a chemical lead are:
In order to decrease the number of compounds that fail in the drug development process, a drug ability assessment is often conducted. This assessment is important in transforming a compound from a lead molecule into a drug. For a compound to be considered druggable it should have the potential to bind to a specific target; however, also important is the compound‘s pharmacokinetic profile regarding absorption, distribution, metabolism, and excretion. Other assays will evaluate the potential toxicity of the compound in screens such as the Ames test and cytotoxicity assay.
Lead Optimization
Lead optimization is the process by which a drug candidate is designed after an initial lead compound is identified. The process involves iterative series of synthesis and characterization of a potential drug to build up a representation of in what way chemical structure and activity are related in terms of interactions with its targets and its metabolism. Leads are further optimised to enhance their efficacy, safety, and pharmacokinetic properties In initial drug discovery, the resulting leads from hit-to-lead high throughput screening tests undergo lead optimization, to identify promising compounds. Potential leads are evaluated for a range of properties, including selectivity and binding mechanisms during lead optimization, as the final step in early stage drug discovery. The purpose of lead optimization is to maintain favorable properties in lead compounds, while improving on deficiencies in lead structure. In order to produce a pre-clinical drug candidate, the chemical structures of lead compounds (small molecules or biologics) need to be altered to improve target specificity and selectivity. Pharmacodynamic and pharmacokinetic parameters and toxicological properties are also evaluated. Labs must acquire data on the toxicity, efficacy, stability and bioavailability of leads, in order to accurately characterize the compound and establish the route of optimization.[15] Researchers in drug discovery need rapid methods to narrow down the selection of drug candidates for this downstream selectivity profiling and further investigation. High throughput DMPK (drug metabolism and pharmacokinetics) screens have become an essential part of lead optimization, facilitating the understanding and prediction of in vivo pharmacokinetics using in vitro tests. In order to make new drugs with higher potency and safety profiles, chemical modifications to the structure of candidate drugs are made through optimization.
Automated screening systems are becoming an important part of pharmaceutical and biopharmaceutical drug discovery labs. Mass spectrometry is used for the detection and quantitation of metabolites. MALDI imaging is a key technique for evaluating drug candidates and their metabolites in tissue structure rapidly and accurately. Additionally, NMR Fragment-based Screening (FBS) in the pharmaceutical industry has become a widely applied method for the discovery and optimization of lead molecules in targeted screening campaigns.[16]
Product Characterization
When any new drug molecule shows a promising therapeutic activity, then the molecule is characterized by its size, shape, strength, weakness, use, toxicity, and biological activity. Early stages of pharmacological studies are helpful to characterize the mechanism of action of the compound.
Formulation and Development
Pharmaceutical formulation is a stage of drug development during which the physicochemical properties of active pharmaceutical ingredients (APIs) are characterized to produce a bioavailable, stable and optimal dosage form for a specific administration route.
During preformulation studies the following parameters are evaluated:
Preclinical Testing
Preclinical research in drug development process involves evaluation of drug safety and efficacy in animal species that conclude to prospective human outcome. The preclinical trials also have to acquire approval by corresponding regulatory authorities. The regulatory authorities must ensure that trials are conducted in safe and ethical way and would give approval for only those drugs which are confirm to be safe and effective. ICH has established a basic guideline for technical necessities of acceptable preclinical drug development.[17] The pre-clinical trials can be conducted in two ways: General pharmacology and Toxicology. Pharmacology deals with the pharmacokinetic and pharmacodynamic parameters of drug. It is essential to explore unwanted pharmacological effects in suitable animal models and monitoring them in toxicological studies. Pharmacokinetic studies are very important to make known the safety and efficacy parameters in terms of absorption, distribution, metabolism and excretion. These studies give information on absorption rate for diverse routes of administration, which helps in selection of dosage form, distribution, rate of metabolism and elimination; which governs the half-life of the drug. Half-life of the drug clarifies the safety outline of the drug which is the obligatory for a drug to get approved by regulatory agencies. The drug distribution mechanism elucidates the therapeutic effectiveness of the drug as it depends on the drugs bioavailability and its affinity. Drug metabolism provides the probability of through phases of biotransformation process and formation of drug metabolites. It also helps in understanding the reactions as well as enzymes involved in biotransformation. [18] Toxicological studies of the drug can be performed by in vitro and in-vivo test which evaluate the toxicological effects of the drug. In-vitro studies can be performed to inspect the direct effects on cell proliferation and phenotype. In-vivo studies can be performed for qualitative and quantitative determination of toxicological effects. As many drugs are species specific, it is essential to select appropriate animal species for toxicity study. In-vivo studies to evaluate pharmacological and toxicological actions, including mode of action, are often used to support the basis of the proposed use of the product in clinical studies.
The Investigational New Drug Process (IND)
Drug developers must file an Investigational New Drug application to FDA before commencement clinical research. In the IND application, developers must include:
Clinical Research
Clinical trials are conducted in people (volunteer)and intended to answer specific questions about the safety and efficacy of drugs, vaccines, other therapies, or new methods of using current treatments. Clinical trials follow a specific study protocol that is designed by the researcher or investigator or manufacturer. As the developers design the clinical study, they will consider what they want to complete for each of the different Clinical Research Phases and starts the Investigational New Drug Process (IND), a process they must go through before clinical research begins. Before a clinical trial begins, researchers review prior information about the drug to develop research questions and objectives. Then, they decide:
Phase 0
Also known as exploratory or piolet studies, phase 0 trials are not mandatory for drug approval and relatively uncommon. Microdosing study This is a new strategy being developed to reduce the cost and time of the drug development process. The rate of rejection of candidate drugs at various stages of clinical development has progressively increased recently, discouraging pharmaceutical companies to venture into the risky business of new drug invention. This has alarmed the FDA (USA) and the European Medicines Agency to encourage novel cost-cutting approaches in drug development. One such tool is the microdosing human study undertaken before phase-1 trial, and is also called phase ‘0’ study
Phase I: Human pharmacology and safety
Phase I trials are the first step in testing an experimental drug or treatment in humans. The first human administration of the drug is carried out by qualified clinical pharmacologists/trained physicians in a setting where all vital functions are monitored and emergency/ resuscitative facilities are available. Sub jects (mostly healthy volunteers, sometimes patients) are exposed to the drug one by one (total 20–80 subjects), starting with the lowest estimated dose (generally 1/100 to 1/10 of the highest dose producing no toxicity in animals) and increasing stepwise to achieve the effective dose. An attempt is made to determine the dose range that may be used in further studies. The emphasis is on safety, tolerability, and to detect any potentially dangerous effects on vital functions, such as precipitous fall/rise in blood pressure or heart rate, arrhythmias, bronchospasm, seizures, kidney/liver damage, etc. Unpleasant side effects are noted and an attempt is made to observe the pharmacodynamic effects in man. The human pharmacokinetic parameters of the drug are measured for the first time. No blinding is done: the study is open label.
Phase II: Therapeutic exploration and dose ranging
Phase II trials are conducted in larger group of patients by physicians who are trained as clinical investigators, and involve 100–500 patients selected according to specific inclusion and exclusion criteria. The primary aim is establishment of therapeutic efficacy, dose range and ceiling effect in a controlled setting. Tolerability and pharmacokinetics are studied as extension of phase I. The study is mostly controlled and randomized, and may be blinded or open label. It is generally carried out at 2–4 centres. The candidate drug may get dropped at this stage if the desired level of clinical efficacy is not obtained. Phase III: Therapeutic confirmation/ comparison Generally these are randomized double blind comparative trials conducted on a larger patient population (500–3000) by several physicians (usually specialists in treating the target dis ease) at many centres. The aim is to establish the value of the drug in relation to existing therapy. Safety and tolerability are assessed on a wider scale, while pharmacokinetic studies may be conducted on some of the participants to enlarge the population base of pharmacokinetic data. Indications are finalized and guidelines for therapeutic use are formulated. A ‘new drug application’ (NDA) is submitted to the licencing authority (like FDA), who if convinced give marketing permission. Restricted marketing permission for use only in hospitals with specific monitoring facilities, or only by specially trained physicians may be granted in case of toxic drugs which are found useful in serious or otherwise incurable diseases.
Phase IV : Postmarketing surveillance/data gathering studies
After the drug has been marketed for general use, practicing physicians are identified through whom data are collected on a structured proforma about the efficacy, acceptability and adverse effects of the drug in the real field situation (similar to prescription event monitoring). Patients treated in the normal course form the study population: numbers therefore are much larger. Uncommon/idiosyncratic adverse effects, or those that occur only after long-term use and unsuspected drug interactions are detected at this stage. Patterns of drug utilization and additional indications may emerge from the surveillance data. Further therapeutic trials involving special groups like children, elderly, pregnant/lactating women, patients with renal/hepatic disease, etc. (which are generally excluded during clinical trials) may be undertaken at this stage. Modified release dosage forms, additional routes of administration, fixed dose drug combinations, etc. may be explored
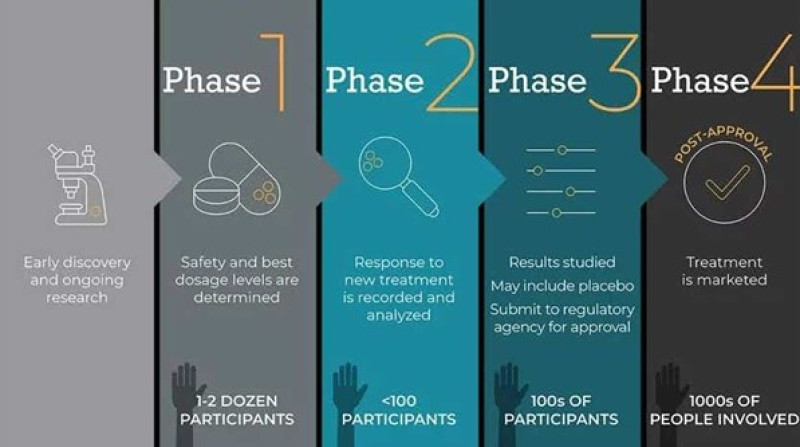
Figure 2 : Phases Of Clinical Trials
REFERENCES


Ganesh U. Chavan, Vitthal. R. Gawade, The Stages Of Drug Discovery And Development Process, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 3, 474-482. https://doi.org/10.5281/zenodo.10603065
 10.5281/zenodo.10603065
10.5281/zenodo.10603065