
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Dr. Vedprakash Patil College of Pharmacy, Georai Tanda
Abrocitinib exists as a Biopharmaceutics Classification System (BCS) Class II drug because it shows poor aqueous solubility and limited oral bioavailability while functioning as a selective Janus kinase 1 (JAK1) inhibitor for treating moderate to severe atopic dermatitis as an oral therapeutic. The research established and optimized a self-emulsifying drug delivery system (SEDDS) for abrocitinib with the purpose of enhancing solubility and dissolution properties. The emulsification analysis indicated almond oil should be used as oil phase while Tween 80 served as surfactant and PEG 400 functioned as co-surfactant because they demonstrated the best emulsification results and solubilization power. The exploration of critical formulation parameters such as emulsification time and percentage transmittance along with drug content involved a 3² full factorial design through modifications of oil and Smix concentrations.
Overview of Abrocitinib
Therapeutic Class and Mechanism of Action
Abrocitinib is a potent oral small molecule that acts as a Janus kinase inhibitor. More specifically, it shows a high selectivity for the Janus kinase isoform one and has become a crucial research tool for inhibiting inflammatory immune-mediated pathways. The Janus kinase family consists of four cytoplasmic tyrosine kinases: Janus kinase one, Janus kinase two, Janus kinase three, and tyrosine kinase 2. Although these kinases are not receptors, they play a critical role in the intracellular signaling cascade that transmits extracellular signals from cytokines and growth factors directly to the nucleus [1].This signaling process is known as the Janus kinase signal transducer and activator of transcription pathway, which plays a crucial role in the growth, differentiation, survival, and proliferation of immune cells. Once the cytokines attach to their specific membrane-bound receptors, the Janus kinases are triggered through auto-phosphorylation.Janus kinases subsequently activate the intracellular domain of the receptor, phosphorylating specific tyrosine residues that act as docking sites for signal transducer and activator of transcription proteins. Following their phosphorylation by Janus kinases, these proteins dimerize, move to the nucleus, and attach to particular DNA sequences to modulate the transcription of target genes involved in immune regulation and inflammatory responses. This pathway is crucial in the development of various autoimmune and inflammatory diseases, including atopic dermatitis [2].
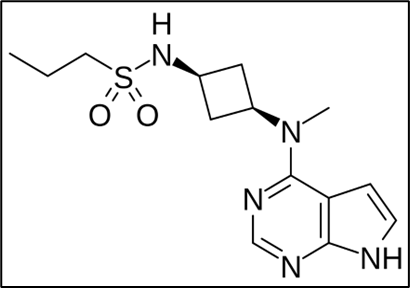
Fig. 1.1 Chemical structure of abrocitinib
These cytokines are recognized for their significant roles in the inflammation process, contributing to the clinical manifestations of atopic dermatitis, such as itching, redness, skin thickening, and lichenification. Abrocitinib functions as a Janus kinase 1 inhibitor, meaning it inhibits the downstream transmission of inflammatory mediator transcription, thereby addressing the molecular underpinnings of the disease's signs and symptoms. Notably, abrocitinib exhibits minimal affinity for Janus kinase 2 and Janus kinase 3, which are involved in signaling for hematopoietic growth factors and interleukins that influence immune cell balance.Like other isoforms, they are often hindered by unwanted hematological and immunological side effects (such as anemia, neutropenia, thrombocytopenia, and a heightened risk of infections). Consequently, abrocitinib exhibits a high selectivity for Janus kinase one, which improves its therapeutic efficacy and results in greater safety and tolerability in comparison to less selective Janus kinase inhibitors [4].
Fig. 1.2 Mechanism and impact of abrocitinib
Introduction to Self-Emulsifying Drug Delivery Systems
Definition and Principle of SEDDS
The self emulsifying drug delivery systems or SEDDS is the superior type of lipid based formulation that has been formulated to increase the oral bio availability of the poorly water soluble drugs. They are isotropic oil, surfactant and co-surfactant or co-solvent mixtures which upon easy agitation in the aqueous phase of the gastrointestinal tract spontaneously form rooms between the oil and water phases to form emulsions in oil or microemulsions without any added energy or force. The SEDDS self emulsification undergoes as a thermodynamically driven transformation process, thus do not require specialized equipment, high shear forces, or any external emulsifying agents, which is usually distinguishing them against conventional emulsion based systems, which entail the emulsification with the utilization of mechanical energy [10]. The essence of SEDDS relies on the fact that further enhancement of the solubilization of the drug in gastrointestinal environment lies in the ability to keep the drug in a solubilized form in lipid drops during the digestion and specific absorption operation. Mild peristatic motions combined with water surroundings are enough to induce dispersion of a SEDDS formula into fine droplets of magnitude nanometer to sub-micro, yet larger than 0.1 inches when introduced to the gastrointestinal system. This is because the emulsified droplets present enormous interfacial surface area within which the drug can diffuse into and as such increases the rate of dissolution and accelerates absorption of the drug into the intestinal epithelia through a swift and effective process [11] Fine emulsions are thus formed which increases the surface area of the lipophilic drug available at the absorption site and neutralizes the slow rate of dissolution which is the main limiting factor of the bioavailability of the Biopharmaceutics Classification System Class 2 drug. They also have the capacity of maintaining the drug in solubilized form as they move through the gastrointestinal tract by the fact that the components of SEDDS and in particular long chain or medium chain triglycerides, non ionic surfactants and hydrophilic co solvents have a concomitant ability to help keep the drug in the solubilized state to avoid one likely result that may otherwise occur when the drug becomes in solubilized form and that is reduction in the drug absorption. One of the most crucial functional properties of SEDDS is maintenance of drug solubility after administration, which allows pharmacokinetics to be subject to more reliable and predictable methods [12]. The type of SEDDS surfactant typically contains a high hydrophilic - lipophilic balance value, to enable the distribution of the lipid phase in aqueous medium and create the stability of the emulsion droplet by reducing the interfacial tension. To enhance the capacity of the system to solubilise further and also to minimize the size of the droplets once the emulsion is made, co-surfactants and co-solvents such as the derivatives of polyethylene glycol or propylene glycol are incorporated. These ingredients and their proportion need to be optimized to prepare an efficient self emulsification with ideal size distribution of the emulsion droplets and which contribute to physical stability of the system [13].Also, solubilization is not the only ancillary mechanism of SEDDS drug absorption process. These lipid elements may affect stimulation of bile secretion, lymphatic clearance, and alterations in intestinal permeability through interaction with membrane lipids or efflux transporters, e.g., P- glycoprotein. These processes increase the overall exposures and reduce the first-pass metabolism of lipophilic, metabolic enzyme or efflux protein drug substrates in the intestinal or liver wall. The compatibility of excipients with the drug substance and solubilizing, and emulsification capacity, all of which have to be done in a systematic manner in the design of an effective SEDDS formulation. In many cases, pseudo-ternary phase diagrams are drawn in order to determine the best proportions of oil, surfactant and co-surfactant in terms of which a stable and effective self emulsifying region can be generated. To push the formulation towards in vivo testing, other vitro testing parameters like emulsification time, droplet sizing analysis, drug release study and thermodynamic stability of the drug is also evaluated [14].
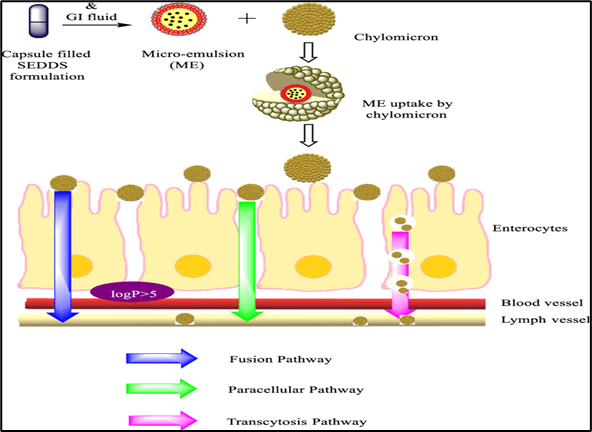
Figure 1.3: Mechanism of SEDDS
Composition: Oil, Surfactant, and Co-Surfactant
A self-emulsifying drug delivery system is composed of three major components: oil, surfactant and co surfactant or co solvent. The interdependency of these components and their contribution to the capacity of solubilization and emulsification efficiency, droplet size distribution, stability and performance of the formulation will be discussed. In particular, the selection and optimization of these components are key for SEDDS development to succeed, especially in the case of abrocitinib, a drug with poor aqueous solubility and high degree of lipophilicity for which the improvement of oral bioavailability is requisite [15].
Oil Phase
The oil component acts not only as the primary medium to solubilize the lipophilic drug but also crucial for governing the self-emulsification process and the obtained droplet size upon dilution. SEDDS can be based on natural, semi synthetic or synthetic triglycerides and divided into long chain triglycerides and medium chain triglycerides according to the chain length of the oil. Drug loading capacity and the capacity of the formulation to form stable emulsions under gastrointestinal conditions greatly depend on the oil used. Commonly used are medium chain triglycerides (MMC), such as the caprylic and capric acid esters, that exhibit better emulsification properties, are rapidly digested and with the gastrointestinal tract. [16].
Surfactant
Surfactants can play an important role by minimizing the interfacial tension between oil and aqueous layers and hence result in spontaneous emulsification when diluted in the gastro intestinal fluids. The reason why Tween 80 (polyoxyethylene sorbitan monooleate) was used as a surfactant in abrocitinib formulation is the outstanding emulsifying activity and high solubilising capacity of poorly water-soluble potentiation, as well as the well-documented safety of this surfactant in oral preparations. Tween 80 has the ability to enhance the dispersibility of lipid droplets and enhance the pace of self-emulsification of the droplets, once exposed to gastric fluids [17].
Co-Surfactant
Co-surfactant or Co-solvent is the other component to increase the extensibility of the interfacial surface film and reduce the depression of surfactant concentration to perform the brookage. It also contributes towards additional suppression of interfacial tension and spreading of surfactant on the surface of oil in order to react more uniformly and quicker with molecules of oil. The co-surfactants are also relevant in improving the solubilization of the drug through enhancing polarity of the formulation as well as stretching the area of self-emulsification within the ternary phase diagram. The popular co-surfactants are short chain alcohols, polyethylene glycol derivatives and propylene glycol derivatives. As the co surfactant in the version shown here polyethylene glycol (PEG) 400 was added due to the hydrophilic nature, satisfactory miscibility with oil and surfactant and relatively simple production of small and stable droplets. PEG 400 is further utilized as a co-solvent which allows the formulation of abrocitinib to dissolve in preparation and it prevents abrocitinib precipitation during GI movement. [18].
Advantages Over Conventional Formulations in Enhancing Solubility and Absorption
Improved Aqueous Solubility
Self-emulsifying drug delivery systems significantly enhance the apparent solubility of poorly water-soluble drugs by forming fine oil-in-water emulsions upon contact with gastrointestinal fluids. This enables the drug to remain in a solubilized state throughout its transit in the gastrointestinal tract, unlike conventional formulations where the drug may remain partially or completely undissolved, limiting absorption [24].
Increased Surface Area for Absorption
The spontaneous formation of nano-sized or micro-sized emulsion droplets greatly increases the interfacial surface area available for drug absorption. This enhanced surface area facilitates a more rapid diffusion of the drug across the intestinal epithelium compared to larger particles or crystalline forms used in conventional tablets and capsules.
By passing the Dissolution Step
In conventional formulations, the drug must first dissolve in gastrointestinal fluids before it can be absorbed. For poorly soluble drugs, this dissolution step is slow and incomplete. SEDDS bypass this limitation by presenting the drug in a pre-dissolved form within lipid droplets, making it immediately available for absorption.
Minimization of Drug Precipitation
Poorly soluble drugs often precipitate upon exposure to aqueous fluids following administration in conventional solid dosage forms. SEDDS stabilize the drug within emulsified lipid droplets, reducing the risk of precipitation even upon dilution or pH changes in the gastrointestinal environment [9].
Reduction in Food Effect
The absorption of many lipophilic drugs can be significantly influenced by food intake due to its effect on bile secretion and gastrointestinal motility. SEDDS minimize food-dependent variability by self-emulsifying efficiently regardless of prandial state, providing more consistent bioavailability compared to food-sensitive conventional formulations.
Improved Lymphatic Transport
Lipid-based systems such as SEDDS can promote the absorption of lipophilic drugs via the intestinal lymphatic system, bypassing hepatic first-pass metabolism. This route can enhance systemic exposure and reduce metabolic degradation, an advantage not typically achievable with conventional oral formulations.
Enhanced Rate and Extent of Absorption
Due to rapid emulsification, improved solubilization, and increased permeability, SEDDS facilitate faster onset of action and higher peak plasma concentrations. In contrast, conventional formulations may exhibit delayed or variable absorption patterns, particularly for Biopharmaceutics Classification System Class Two drugs.
Mechanistic Benefits of SEDDS in Oral Delivery
Self-emulsifying drug delivery system is the latest development of the pharmaceutical technology particularly in poorly water soluble orally delivered drugs. The mechanistic benefits of such systems are entirely unprecedented in novelties beyond solubilizations. They are based upon physicochemical or physiological considerations in the gastrointestinal tract and are developed in order to have a more favorable dissolution behavior, a better permeation and more predictable systemic exposure. It is at this point that the emulsification process is fast and effectively efficient with mild gastrointestinal motility that includes peristaltic motion, churning, etc., on these foods to produce emulsions of droplet sizes which varies between a few nanometers to submicrometer dimensions [8]. Because of high interfacial surface area of these nano or micro sized droplets, the partitioning rate of the drug into the liquid aqueous phase is raised. Greater surface area provides greater intimacy between intestinal epithelium and any marginated drug containing lipid droplet and therefore a faster and large absorption. This mechanism is especially helpful with lipophilic drugs that would otherwise be subject to mixing in the gut concretely and partially in the classical dosage forms. High surface area of the donors also blends the process of dissolution and ensures a constant concentration gradient with respect to drug diffusion through the biological barrier. Among the major mechanical benefits of SEDDS, is their appeal of preventing the precipitation of the drug once it has been released into their gastrointestinal transit. The hydrophobic drug may be dissolved in huge amounts inside the oil-stage (generally, long chain or middle chain triglycerides) and generate a lipophilic environment. In addition, the emulsified drops are even more stabilized by surfactants and co surfactants especially through the establishment of the strong interfacial film that inhibit the coalescence of these drops and the precipitation of the drug. To prevent generating issues in the stomach or the intestine, this protective mechanism is necessary to make sure that the drug stays as a solution in various pH conditions of the intestine and stomach. Unlike conventional formulations SEDDS based formulations can withstand pH changes, dilution and exposure to bile salts and still solubilize drugs in difficult physiological conditions [9]
LITERATURE REVIEW
Gao et al. (2021) In-depth research on the designing and evaluation of a self-emulsifying drug delivery system (SEDDS) of cepharanthine (CEP) to enhance its oral bioavailability was reported. They carried out solubility screening and pseudo ternary phase diagram analysis of select oil phase (isopropyl palmitate), surfactant (Cremophor RH40) and co surfactant (Hypromol 500) and the solubility screening demonstrated that the three combinations formed are in accordance with what existed in literature. An optimal formulation with a CEP:IPP:Cremophor RH40:PEG 200 ratio was 3.6:30.0:55.3:11.1 (w/w) (resultant) and this mixture was optimized through a central composite design under response surface methodology, and produced a high drug loading (36.21 mg/mL) and low particle size (36.70 nm). Analyses of dynamic light scattering, transmission electron microscopy and in vitro release are carried out to discern the optimized composition. In buffered phosphate solution (pH 6.8) the CEP-SEDDS liberated about 100 percent of drugs within less than half an hour whereas pure CEP liberated roughly 20 percent. Its in vivo pharmacokinetics in rat after oral administration was also identified as having relative bioavailability of 203.46% [2].
Cardona et al. (2021) In this article they documented the formulation and optimization of an orally bioavailable self-emulsifying drug delivery system (SEDDS) of calyces extract of Physalis peruviana to achieve the enhancement in oral bioavailability of the active flavonoids of this extract and in particular rutin and hypoglycemic activity of the active flavonoids of this extract as well. Constant solubility tests were carried out to choose ingredients of SEDDS Labrafac to be used as oil (Labrafac), surfactant (Solutol HS 15) and co-solvent (propylene glycol). The optimization was conducted within the Box-Behnkan statistical design using an ideal mixture of Labrafac 10 percent, Solutol HS 15 45 percent, propylene glycol 32 percent as well as PDMSHEPMS 13 percent (all w/w) with the chosen extract at 45 percent (w/w). The SEDDS was optimized as indicated by a mean size of the droplet of 19.5 nm, polydispersity index (PDI) of 0.20 and negative zeta potential but is well formed. Permeation studies using Transwell mucus were also carried out; the unformulated extract and consequent values of permeation were recorded at 2.6-fold higher than the unformulated extract. Oral bioavailability of rutin was also increased by almost 6-fold in a pharmacokinetic analysis. Additionally, the extract loaded SEDDS showed higher activity of the extract in hypoglycemic activity than that of pure extract, hence, suggesting that the extract loaded SEDDS could be used as a means of enhancing therapeutic effect of plant-derived therapeutics like flavonoids [4].
Silva et al. (2024) An anti-fungal drug RN104, a thiazolylhydrazone derivative was found to have good antifungal activity and a self-emulsifying drug delivery system SEDDS was developed and biopharmaceutical/and oral bioavailability was enhanced with it. The formingulation components that are medium chain triglycerides, sorbitan monoleate and polysorbate 80 were selected based on solubility studies and construction of pseudoternary phase diagram. SEDDS formulation was characterized by using dynamic light scattering, in vitro drug release, polarized light microscopy, transmission electron microscopy, and stability studies. There was a marked increase in an oral bioavailability (2133%) relative to the free drug, as measured by the pharmacokinetic in vivo assessment done on mice of RN104-SEDDS. Such results have endorsed the employ of SEDDS as a delivery mode to enhance the oral administration and therapeutic benefits of RN104 as a new antifungal agent [5].
Meirinho et al. (2022) In a comprehensive review, the prospect of self-emulsifying drug delivery systems (SEDDS) in improving the brain bioavailability of poorly water soluble neurotherapeutics following administration via the intranasal route was brought to fore. According to the description, SEDDS are isotropic blends of oils, surfactants and co-surfactants that have the ability to mix with water and produce either nanoemulsion or microemulsion, hence enhancement of solubilization capacity of drugs, permeability of membranes and stability of labile drugs to enzymation. In this review, this level of rimetic insight of SEDDS effect in the nasal cavity was provided with the capacity to inhibit drug precipitation and augmentation of mucosal drug absorption. The in vitro results were also compared to SEDDS because the intranasal administration provided a better brain targeting and even improved the systemic bioavailability relative to traditional formulations in vivo [6].
Asghar et al. (2022) On the basis of this, the preparation and characterization of a self-emulsifying drug delivery system (SEDDS) of ciprofloxacin (CPN) was reported as a solution aimed at enhancing its water solubility and oral availability in addition to its antibacterial effect. Subsequently the pseudo-ternary phase diagrams were analysed to define the most preferential emulsification area with different oils, surfactants and co surfactants to generate eight formulations (F1F8). The best of these was F5-CPN i.e. 40, 60 and 6:1, of oil, polysorbate-80 and propylene glycol respectively. This possessed thermodynamic stability, 202.6 nm droplet size, zeta potential of - 13.9 mV and polydipserity index of 0.226. No drug-excipient interactions were found with the use of FT-IR spectrum and SEM imaging showed consistently spherical droplets [7].
AIM AND OBJECTIVES
Aim
The objective is to develop and enhance a self-emulsifying drug delivery system (SEDDS) for abrocitinib, aiming to improve its solubility in water, enhance its dissolution rate, and boost its oral bioavailability.
Objectives
DRUG AND EXCIPIENTS PROFILE
Table :1.1 Drug- Abrocitinib
|
Synonym |
PF-04965842 – Often referenced in clinical studies and regulatory filings under this designation. |
|
IUPAC Name |
N-(3-((1R,2R)-2-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)cyclopropyl)phenyl)-2-(1H-pyrazol-4-yl)acetamide |
|
Description |
A potent and selective inhibitor of JAK1 that interferes with cytokine signaling pathways. It is designed to reduce inflammatory responses by modulating immune cell activity. Its formulation challenges arise due to poor aqueous solubility, which is being addressed by advanced drug delivery systems such as SEDDS. |
|
Molecular Formula |
C????H??N?O |
|
Molecular Weight |
351.4 g/mol |
|
Chemical Structure |
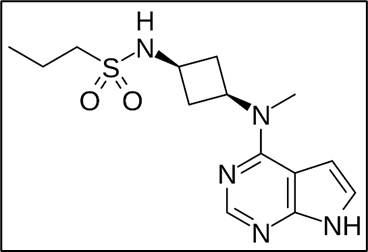
|
|
Solubility |
Classified as a BCS Class II compound with poor aqueous solubility, necessitating formulation strategies like SEDDS to enhance dissolution and bioavailability. The lipid-based delivery system helps to bypass solubility limitations by encapsulating the drug in oil droplets. |
|
pH |
As a pure solid, abrocitinib does not have an intrinsic pH. However, when formulated, the pH of the solution is influenced by the excipients and can be adjusted to optimize drug stability and absorption in the gastrointestinal tract. |
|
Melting Point |
Approximately 150–153 °C – This melting range is used to characterize purity and to inform processing conditions during formulation development. |
|
Mechanism of Action |
Inhibits the JAK1 enzyme, thereby blocking downstream STAT signaling. This interruption in cytokine signaling prevents the transcription of inflammatory mediators, reducing the overall inflammatory response and providing therapeutic benefit in immune-mediated conditions. |
|
Applications |
Primarily used in the management of moderate-to-severe atopic dermatitis. Research is ongoing into its potential application in other inflammatory and autoimmune disorders due to its targeted mechanism of action. |
EXCIPIENTS
Table: 1.2Almond oil
|
Synonym |
Sweet Almond Oil – Commonly known by this name in cosmetic, pharmaceutical, and food industries. |
|
Background |
Extracted from the kernels of Prunus amygdalus, almond oil is a natural vegetable oil renowned for its emollient properties. It has been traditionally used for skin care and is gaining acceptance as a lipid phase in pharmaceutical formulations, especially in enhancing the bioavailability of lipophilic drugs. |
|
Solubility |
Insoluble in water but fully miscible with other oils and non-polar solvents. Its lipophilic nature makes it an excellent candidate for use as the oil phase in SEDDS formulations, enhancing drug solubilization and absorption. |
|
Boiling Point |
Almond oil does not have a typical boiling point as it decomposes upon heating; however, it is thermally stable up to moderate temperatures, making it suitable for formulation processes that do not involve extreme heat. |
|
Melting Point |
Liquid at room temperature (generally remains fluid below 10?°C), which facilitates ease of handling and formulation into emulsions. |
|
Viscosity |
Exhibits moderate viscosity, which is ideal for blending with other formulation components. Its rheological properties allow for uniform mixing and controlled release in drug delivery systems. |
|
Mechanism of Action |
Functions as an inert lipid vehicle that enhances the solubility of lipophilic drugs. It facilitates improved absorption by promoting the formation of a fine emulsion in gastrointestinal fluids. |
|
Applications |
Widely used in topical and oral formulations, especially in lipid-based delivery systems like SEDDS, and also used in cosmetic products for its moisturizing properties. |
Table : 1.3 Tween 80
|
Tween?80 [101,102] |
|
|
Synonym |
Polysorbate?80, Polyoxyethylene (20) sorbitan monoleate – A common nonionic surfactant used in numerous pharmaceutical and cosmetic products to stabilize emulsions. |
|
Molecular Formula |
C??H???O?? |
|
Molecular Weight |
1310 g/mol |
|
Chemical Structure |
Comprises a sorbitan backbone esterified with oleic acid and linked to polyoxyethylene chains. This structure confers both lipophilic and hydrophilic properties, enabling it to effectively stabilize oil droplets in aqueous media. |
|
Solubility |
Highly soluble in water and miscible with many organic solvents, which is beneficial for its role in dissolving both polar and non-polar substances in formulations. |
|
pH |
Typically neutral in aqueous solutions, ensuring that it does not interfere with the pH-sensitive stability of active pharmaceutical ingredients. |
|
Melting Point |
Remains liquid at room temperature, making it easy to handle and incorporate into formulations without the need for melting or other preparatory steps. |
|
Mechanism of Action |
Works by reducing the interfacial tension between oil and water, leading to the formation of a stable emulsion. It stabilizes the interface through steric hindrance, which prevents coalescence of the dispersed droplets. |
|
Stability and Storage Conditions |
Stable at room temperature when stored in a sealed container; it should be kept away from extreme temperatures and reactive chemicals to prevent degradation. Its shelf-life is generally long when maintained under recommended conditions. |
|
Applications |
Extensively used in pharmaceutical, cosmetic, and food products as an emulsifier, solubilizer,and stabilizer. In SEDDS, it plays a key role in improving the dispersion and absorption of poorly water-soluble drugs. |
Table: 1.4 PEG400
|
PEG?400 [103,104] |
|
|
Synonym |
Polyethylene Glycol 400 – Commonly known as PEG?400, it is widely used as a solvent and co-surfactant in various pharmaceutical formulations. |
|
Description |
A clear, low-viscosity, water-miscible liquid that acts as an excellent solubilizer and co-surfactant. PEG?400 improves the miscibility of formulation components and aids in the formation of stable nanoemulsions, making it a critical component in SEDDS formulations designed to enhance drug bioavailability. |
|
Molecular Formula |
H–(O–CH?–CH?)?–OH |
|
Molecular Weight |
400 g/mol |
|
pH |
Typically neutral when in solution, which helps maintain the stability of pH-sensitive drugs and excipients in formulations. |
|
Boiling Point |
Approximately 250 °C; however, PEG?400 tends to decompose before reaching a true boiling point. This thermal behavior is taken into account during formulation development to ensure stability. |
|
Melting Point |
Approximately -65 °C – As a liquid at room temperature, it remains fluid under normal storage conditions, which is advantageous for its incorporation into liquid formulations. |
|
Viscosity |
Exhibits low-to-moderate viscosity, which enhances its ability to act as a solvent and co-surfactant. This property facilitates ease of processing and improves the overall emulsification process in SEDDS, ensuring a uniform distribution of the active drug. |
|
Mechanism of Action |
Acts as a co-surfactant by reducing interfacial tension between the oil and aqueous phases. This promotes the formation of a more stable and homogeneous emulsion, facilitating improved drug dispersion and absorption. |
|
Applications |
Widely used in pharmaceutical formulations, particularly in SEDDS, to improve the solubility and bioavailability of poorly water-soluble drugs. Also employed in cosmetics and other industries where solvent properties are essential. |
MATERIALS AND METHODS
|
Sr. No. |
Name of Chemical |
Purpose of Use |
Manufacturer / Supplier |
|
1 |
Abrocitinib |
Active pharmaceutical ingredient |
Clearsynth Labs Pvt. Ltd., Mumbai |
|
2 |
Almond Oil |
Oil phase in SEDDS formulation |
VedaOils, Delhi |
|
3 |
Tween 80 |
Surfactant in SEDDS formulation |
Loba Chemie Pvt. Ltd., Mumbai |
|
4 |
PEG 400 |
Co-surfactant in SEDDS formulation |
Merck Specialities Pvt. Ltd., Mumbai |
|
5 |
Distilled Water |
Solvent and dilution medium |
In-house Laboratory Supply |
|
6 |
Methanol |
Solvent for HPLC and solubility studies |
Rankem (Avantor), India |
|
7 |
Sodium Hydroxide |
pH adjustment in buffer preparation |
Qualigens Fine Chemicals, Mumbai |
|
8 |
Potassium Dihydrogen Phosphate |
Buffer system component |
Loba Chemie Pvt. Ltd., Mumbai |
|
9 |
Hydrochloric Acid |
Simulated gastric fluid preparation |
SD Fine-Chem Ltd., Mumbai |
Evaluation Properties
Organoleptic properties
A small quantity of the pure drug was placed on a clean, dry watch glass and visually inspected under daylight conditions for its color and appearance. The odor was assessed by gently wafting the sample towards the nose, and any distinct smell was noted. The texture of abrocitinib was determined by carefully rubbing a small amount of the powder between the fingers to observe its physical feel.
|
Sr. No. |
Parameter |
Observed value |
Reported value |
|
1 |
Colour |
white to off-white powder. |
white to off-white powder. |
|
2 |
Odor |
odorless |
odorless |
|
3 |
Texture |
fine powder |
fine powder |
Solubility studies
Different oils were assessed to determine the oil phase that will provide a suitable solvent for abrocitinib in the SEDDS formulation. 2 mL of each oil selected were placed into separate glass vials and excess abrocitinib (5 µlg−1) was added to each. For examinations at equilibrium treatment, the vials were sealed and shaken at 37 ± 0.5°C by periodic orbital shaker (Remi, Model CIS-24BL, India) at an incidence of 100 rpm for 48 hours. Undissolved drug was separated from the samples after incubation by centrifuging at 5000 rpm for 15 minutes (Remi, Model R-8C, India). Supernatant was recovered, suitably diluted with methanol and was analyzed at 288 nm by UV-Visible spectrophotometer (Shimadzu UV-1900, Japan). Using this information, abrocitinib solubility in each oil was calculated as mg/mL. The experiment was carried out in triplicate (n=3) for reproducibility. The same procedure was used for the evaluation of solubility in multiple surfactants and co surfactants [11,12].
|
Components |
Solubility (mg/mL) |
|
Oils |
|
|
Almond oil |
14.72 ± 0.75 |
|
Isopropyl myristate |
11.25 ± 0.58 |
|
Isopropyl palmitate |
9.86 ± 0.51 |
|
MCT |
8.54 ± 0.47 |
|
Captex® 355 |
7.32 ± 0.43 |
|
Ethyl oleate |
6.28 ± 0.39 |
|
Surfactants |
|
|
Brij-30 |
26.74 ± 1.08 |
|
Tween 80 |
30.15 ± 1.25 |
|
Tween 20 |
21.78 ± 0.92 |
|
Span 80 |
17.64 ± 0.83 |
|
Cosurfactants |
|
|
PEG 400 |
39.45 ± 1.51 |
|
Transcutol P |
34.92 ± 1.30 |
|
Propylene glycol |
30.26 ± 1.17 |
|
Ethanol |
26.54 ± 1.00 |
Melting point determination
The melting point of pure abrocitinib was determined to confirm its thermal characteristics. A small quantity of the drug was placed into a capillary tube sealed at one end and introduced into a digital melting point apparatus (Labtronics, Model LT-800D, India). The temperature was gradually increased at a rate of 1–2°C/min, and the temperature at which the drug showed the first sign of melting and the temperature at which it completely liquefied were recorded. The melting point was noted as a range between the onset of melting and complete liquefaction. Each measurement was performed in triplicate (n=3) to ensure accuracy and reproducibility [114,115].
|
Drug Name |
Drug Name |
|
|
Melting Point(ºC) |
Standard |
Observed |
|
149-153ºC |
150-153±1ºC |
|
pH determination
The pH of all prepared abrocitinib-loaded self-emulsifying drug delivery system (SEDDS) formulations was evaluated to assess the physiological compatibility of the emulsified systems upon oral administration and to monitor potential changes during formulation development. As the SEDDS are anhydrous preconcentrates, the pH was determined after dispersion to simulate in vivo gastrointestinal conditions. For each batch, 1 mL of the formulation was diluted with 100 mL of double-distilled water under gentle magnetic stirring to form a uniform emulsion. The pH of the diluted emulsion was measured using a calibrated digital pH meter (Eutech Instruments, Model pH 700, India) maintained at room temperature (25 ± 2 °C). Prior to each measurement, the electrode was rinsed thoroughly with distilled water to prevent cross-contamination. The pH was measured in triplicate (n = 3) for each batch, and the mean values were recorded. These measurements aided in assessing formulation stability and predicting potential mucosal tolerability upon oral administration [25,26]
Self-emulsification efficiency
The efficiency of self-emulsification in the self-emulsifying drug delivery system (SEDDS) was evaluated through a standardized disposability test. This test aimed to assess the formulation's capacity to spontaneously emulsify when it comes into contact with aqueous media under mild agitation, thereby simulating in vivo gastrointestinal conditions. The evaluation was conducted using a USP Type II dissolution apparatus (Electrolab, Model TDT-08L, India), which was filled with 500 mL of distilled water maintained at a temperature of 37 ± 0.5°C, and stirred with a paddle rotating at 50 revolutions per minute (RPM). One milliliter of the SEDDS formulation was introduced into the vessel using a glass pipette that was positioned just beneath the surface of the medium. The emulsification behavior was visually monitored and classified based on clarity, dispersion rate, and the presence of any globules or phase separation. Formulations were evaluated according to a modified dispersibility classification system: Grade A for the rapid formation of a clear or slightly bluish emulsion in less than one minute, Grade B for emulsion formation within two minutes with slight turbidity, Grade C for slow emulsification taking more than two minutes with an opaque appearance, and so on. Each formulation was tested in triplicate (n = 3) to ensure reproducibility [11].
Viscosity
Viscosity measurement was performed to determine the rheological behavior of the self-emulsifying drug delivery system (SEDDS), which is critical for evaluating flow properties, capsule filling suitability, and pourability. The viscosity of each formulation was measured using a digital rotational viscometer (Brookfield, Model DV-E, USA) equipped with spindle number RV-6, operated at a constant speed of 100 revolutions per minute (RPM) at 25 ± 0.5°C. Prior to measurement, the SEDDS formulation was equilibrated at room temperature for 30 minutes to eliminate any temperature-induced variability. Approximately 20 mL of the formulation was transferred into the viscometer beaker, ensuring the spindle was completely immersed without introducing air bubbles. The reading was recorded once a stable torque value was achieved, and viscosity was expressed in centipoise (cP). Each sample was tested in triplicate (n = 3), and results were expressed as mean ± standard deviation (SD). The type of flow (Newtonian or non-Newtonian) was determined based on torque stability and visual consistency during rotation [13].
|
Batch |
Viscosity (cP) |
|
DF1 |
82.6 ± 2.15 |
|
DF2 |
87.4 ± 2.08 |
|
DF3 |
91.2 ± 2.41 |
|
DF4 |
85.1 ± 1.93 |
CONCLUSION:
This study can conclude from the findings that the formulation and optimization of a SEDDS to deliver abrocitinib is definitely an effective solution to the solubility and bioavailability problems encountered with this drug having poor water solubility. Almond oil served as a lipid carrier which allowed the use of a natural and pharmaceutically acceptable vehicle and excellent drug solubilization capacity. By using Tween 80 and PEG 400 as surfactant and co-surfactant, it was rapidly formed fine nanoemulsion upon contact with aqueous media, enabling speed dissolution and absorption in gastrointestinal tract. Because of that, we implemented then 3² factorial design analyzed via a quadratic response surface model which established a robust framework for the optimization process. The optimal concentration of oil and Smix for desired performance, as well as meaningful interactions between formulation variables, were identified by this statistical model, and rational design of the delivery system was enabled. The optimized SEDDS formulation could achieve a fine balance in rapid emulsification, high drug content, and optical clarity, and has an improved in vitro drug release profile compared to the existing formulation. Hence confirmed through accelerated storage studies, the physical stability of the formulation supports this system for long term use as a pharmaceutical. The improved solubility and dissolution behavior as studied constitute a valuable platform to narrow down in vivo target spaces and facilitate further clinical translation of the drug, both based on the assumption that enhanced bioavailability and therapeutic efficacy are to be expected in patients. Overall, it demonstrated that lipid based drug delivery systems (including SEDDS) are promising and scalable approaches to the delivery of poorly solubilized drugs, such as abrocitinib, and made a meaningful contribution towards progress of oral delivery technologies.
REFERENCES


Vaykos Divya K., Dipali Hamde, To Formulation and Optimization of Self-Emulsifying Drug Delivery Systems of Abrocitinib, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 2462-2477. https://doi.org/10.5281/zenodo.16030592
 10.5281/zenodo.16030592
10.5281/zenodo.16030592