
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
B. Pharmacy, Pravra Rural College of Pharmacy, Loni, Maharashtra, India-413736
Ensuring Quality in Pharmaceutical Manufacturing should follow ISO9001:2015 Standards and guidelines from regulatory bodies like the FDA, EMA, WHO. To ensure product quality, safety and efficacy which require process validation to guarantee the quality of their products. This applies to both finished drugs and active pharmaceutical ingredients (APIs). The Goal of Quality is the primary objective of Quality Control can produce medications that are secure and essential for their intended use. Process validation involves collecting and analyzing data throughout the manufacturing process, from design to production, to confirm that it consistently produces high quality products.
The USFDA’s 2011 Process Validation Guidelines outline three key stages:
3.Continued Process Verification – Ongoing monitoring to ensure consistent product quality. Before a product can be sold, manufacturers must gather enough data from product development, scale-up studies, equipment testing, and initial production batches to prove the process is reliable. superior products. [3] The purpose of the USFDA's pharmaceutical product guidelines is to covenant and medications are premium, secure and reliable. These initial batches, known as conformance batches, confirm that the commercial-scale process works correctly under set conditions. A well-validated manufacturing process ensures a high level of confidence in producing consistent, high-quality products [3] The USFDA guidelines for pharmaceutical products are designed to make sure such drugs are superior, productive, capable, and of best quality. These guidelines cover various stages of drug development, manufacturing, and post-market surveillance. Below are the key components 1. Drug Development and Approval Preclinical Testing: Before testing humans, pharmaceutical companies must conduct animal studies to evaluate the safety and potential risks. Investigational New Drug (IND) Application: To begin clinical trials in humans, the company must submit an IND application, including results of preclinical studies, a proposed clinical trial plan, and the manufacturing information. Clinical Trials: [4,5]
Phase 1: Safety and dosage testing in healthy volunteers (20-100 individuals).
Phase 2: potency and adverse reaction testing in small patient population with the circumstances (100-300 individuals)
Phase 3: Extensive trials to adhere effectuality, observe outcome, and differentiate with excellence treatments (1,000-3,000 individuals).
Phase 4: Post-market studies to monitor long-term safety, efficacy, and new uses. New Drug Application (NDA) to the U.S. Food After and Drug Administration contains compressive data from pre-clinical and clinical studies. The FDA thoroughly reviews the NDA to determine whether the drug should be approved for public use. The FDA evaluates the submitted information collected to confirm the drug’s safety effective, manufactured according to high standards. If approved, the drug can be marketed. [6]
1.FDA enforces Current Good Manufacturing Practices:
(CGMP) to guarantee that drugs are manufactured to and adhere to quality standards. This includes:
Facility Standards: Ensures that facilities used for production are clean and well-maintained. Personnel Requirements: Manufacturers must employ qualified personnel, and training should be ongoing. Quality Control and Testing: The manufacturing process should be documented and regularly reviewed. Finished products must be tested for protection, cleanliness, and strength.
Batch Records with Documentation: Manufacturers must maintain detailed records of particular batch of drug produced to assure traceability and accountability. [1,7]
2.Drug Labelling Requirements:
The FDA mandates that pharmaceutical products must have clear, accurate, and comprehensive labelling, which includes: Brand and Generic Names. Active Ingredients and their concentrations. Indications (conditions the drug treats). Dosage Information (how to take the drug, recommended dosages). Warnings and Precautions (e.g., contraindications, side effects, drug interactions). Storage instruction, expiration, date manufacturer Information. [8]
3. After a drug is approved: ongoing monitoring is essential:
Adverse Event Reporting: The FDA requires pharmaceutical companies to report any adverse reactions, side effects, or new safety issues. FDA Adverse Event Reporting System (FAERS): This system collects reports of adverse events to identify potential safety issues with marketed drugs. Risk Evaluation and Mitigation Strategies (REMS): If necessary, drugs with known serious risks are subject to REMS to ensure safe use. This may include restricted distribution, patient education, or specific monitoring. [9]
4.Drug Recalls:
The FDA can request or enforce a recall of pharmaceutical products if they are found to be unsafe, ineffective, or non-compliant with regulations. There are three classes of recalls based on the risk to public health:
Category 1: Hazardous or faulty products that present a significant health risk.
Category 2: Items that could lead short-term or permanent health complications.
Category 3: Items that are not expected to result in harmful health effects. [10]
5.Advertising and Promotion:
The FDA oversees the marketing and advertising of pharmaceutical products to ensure that all claims made are truthful, not misleading, and substantiated by scientific evidence. This includes:
Direct-to-Consumer Advertising (DTCA): The FDA monitors ads that are directed toward consumers to ensure they are accurate and contain necessary information about the risks and benefits of a drug. Off-Label Promotion: Pharmaceutical companies are prohibited from promoting drugs for uses not approved by the FDA. Reference to the use on the label for indications not included in the approved labelling.
The FDA offers various programs accelerated authorization for the approval of certain medicines:
Fast Track: Accelerates the review the procedure for medications that address severe conditions and fulfill an unmet medical necessity.
Priority Review: Shortens the review period for drugs that show significant improvements over existing treatments.
Breakthrough Therapy: Provides more intensive FDA guidance to help develop drugs that offer substantial improvements for serious or life-threatening conditions.
Orphan Drug Designation: Provides incentives for the development of drugs to treat rare diseases, including tax credits and seven years of market exclusivity.
The FDA also regulates biologic products (such as vaccines, gene therapies, and monoclonal antibodies) under the Public Health Service Act (PHSA). Biologics are typically regulated differently than traditional drugs and require a Biologics License Application (BLA) for approval.
Generic drugs must be proven to be bioequivalent to the branded version. This means they must contain the identical active ingredient, dosage form, strength, and method of administration, and provide the same therapeutic effect. Abbreviated New Drug Application (ANDA): Generic manufacturers submit an ANDA to demonstrate that their drug is bioequivalent to the brand-name medication. Instead of conducting new clinical trials, they are only required to perform bioequivalence studies.
The FDA enforces strict regulations for conducting clinical trials in accordance with Good Clinical Practice (GCP). These guidelines ensure ethical standards are met, safeguard participants rights, and ensure the reliability of the collected data.
The FDA regulates the importation of drugs into the United States to ensure that foreign drugs meet the same standards as those manufactured domestically.
The FDA works with other federal agencies, foreign governments, regulatory coalitions’ (e.g., European Medicines Agency (EMA)to harmonize drug development and approval processes, making it easier for companies to submit drug data globally and ensure consistent standards across countries. These guidelines ensure that pharmaceutical products are developed, manufactured, and marketed in ways that protect public health, ensure that drugs are effective, secured, and adequately regulated throughout their lifecycle. [11]
Fig.1. Flow chart of Food and Drug Administration.
ADVANTAGES:
Laws such as Good Manufacturing Practices (GMP) ensure that pharmaceuticals are consistently produced and handled in according with quality standards. This reduces variability and ensures product homogeneity.
The credibility of pharmaceutical products is increased worldwide by USFDA approval; many countries accept or employ USFDA criteria for medication evaluation, which makes it easier for companies to enter other markets.
When a medication is approved by the USFDA, it means that it meets stringent safety and effectiveness requirements. As a result, the public has more faith in drugs.
Are encouraged to invest in research and development (R&D) with increased confidence in regulatory outcomes since USFDA guidelines offer a clear framework for drug development and clinical trials.
Guidelines guarantee that medicine labels accurately convey information concerning warnings, dosage, side effects, and indications. Patients and healthcare professionals can use this to make well-informed decisions.
7.Reduces Litigation and Recall Risk:
Following USFDA guidelines lessens the likelihood of product recalls and legal issues resulting from safety-related issues. This protects patients and pharmaceutical companies against potential harm and financial loss.
To monitor safety concerns and adverse events, the USFDA monitors drugs after they are placed on the market. This makes it possible to act quickly in the event that new threats are identified.
Products that adhere to USFDA standards typically have a competitive edge over rivals who don’t for businesses that produce FDA-compliant items, this gives them a competitive advantage. [14]
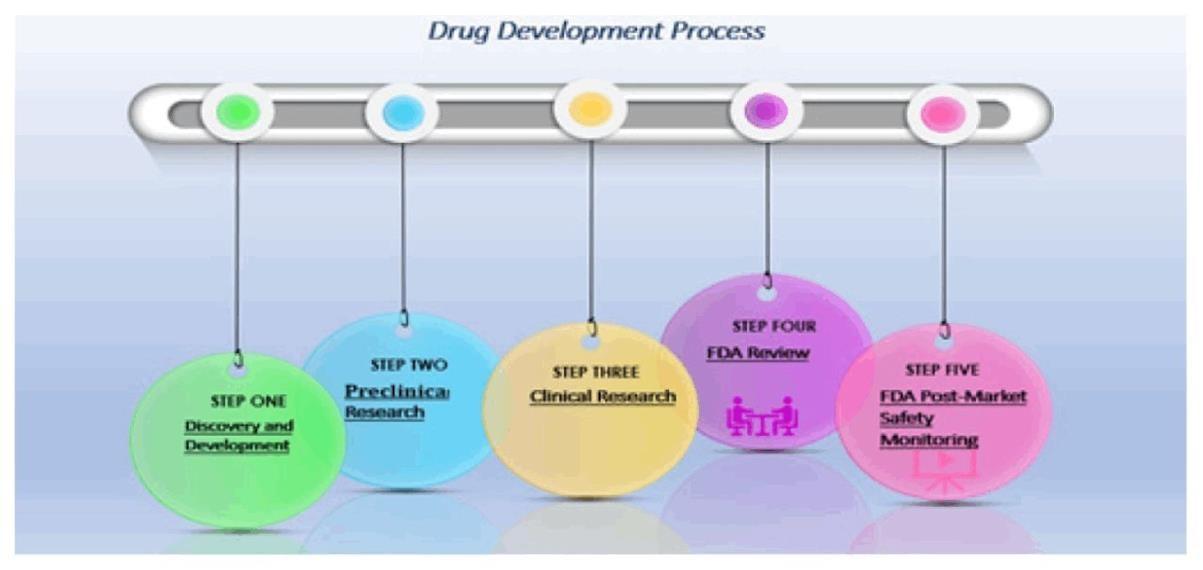
Fig.2.Flow chart of Drug Development Process.
To ensure the safety and effectiveness of new drugs, the drug development process consists of multiple steps, including discovery and research, preclinical studies, clinical trials, regulatory assessment, and post-market safety monitoring. Finding viable treatment candidates and comprehending the fundamental causes of illnesses are the main goals of this first phase. Lead Compound Identification: Scientists find chemicals that show promise in treating a certain illness. Verifying that the identified target is, in fact, a suitable treatment target is known as target validation. Mathematical Optimization: Drug design can be optimized and drug behavior can be predicted using mathematical models.
Pharmacokinetics and Drug Disposition: Successful drug development requires an understanding of how a drug is absorbed, transported, metabolized, and eliminated.
Pre-formulation Development: This phase concentrates on the drug's physicochemical characteristics and appropriateness for dosage form formulation. [14]
Usfda Inspections Database:
This database should not be used to gather official data because it is not an exhaustive record of all inspections that have been carried out. This excludes state-conducted inspections, pre-approval inspections, inspections of nonclinical labs, inspections of mammography facilities, and inspections pending a final enforcement action. Nonclinical Laboratories Inspected under Good Laboratory Practices has inspection data sets for nonclinical laboratories. [12]
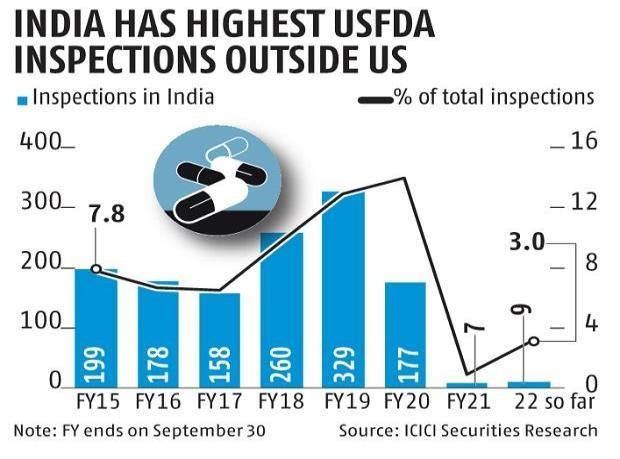
Fig.3.Graph of USFDA Inspections.
DISCUSSION:
The USFDA authorities are important for maintaining public health since it regulates pharmaceutical products. This includes the creation, production, by guaranteeing that pharmaceutical items are continuously manufactured and regulated in accordance with quality standards, FDA regulations aim to safeguard the public's health. Adhering to these guidelines lowers the risk of contamination, incorrect labelling, ineffective dosage, and adverse effects. By following these guidelines, pharmaceutical companies can get FDA approval, avoid problems, and maintain consumer trust. Following FDA guidelines is not only mandated by law, but it is also crucial for the distribution of pharmaceuticals and for ensuring that patients receive safe and effective medications. To ensure compliance and uphold quality standards, the FDA issues a variety of recommendations and regulations.
Documents with FDA Guidance:
The FDA's current stance, as stated in guidance papers, on specific matters related to drug research and manufacture. These documents provide the industry with useful recommendations and insightful analysis, even though they are not legally binding. Stakeholders can search for these guidelines using keywords and filter results by product type, FDA organizational unit, date of issue, document type, subject, draft or final state, and comment period dates. [13]
CONCLUSION:
The FDA in the United States establishes and enforces regulations to ensure the quality, safety, and effectiveness of pharmaceutical products. These regulations cover every phase of the drug lifecycle, including research and development, manufacturing, labelling, post-market surveillance and marketing. The primary aim of FDA guidelines are is to protect public’s health by ensuring that pharmaceutical products are consistently produced and meet strict quality standards. Complying with these rules helps reduce the risk of contamination, incorrect labelling, improper dosage, and adverse reactions. By following these guidelines, pharmaceutical companies can get FDA approval, avoid problems, and maintain consumer trust. Following FDA guidelines is not only mandated by law, but it is also crucial to giving patients safe and efficient medications.
REFERENCES






Dr. S. D. Mankar, Kalyani Nikam*, Vaishnavi Palve, Vaishnavi Nirmal, Pallavi Palve, Jagdish Nikam, USFDA Guidelines: A review on Pharmaceuticals Formulations, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 4, 1473-1479. https://doi.org/10.5281/zenodo.15201903
 10.5281/zenodo.15201903
10.5281/zenodo.15201903