
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutics, Nirmala College of Pharmacy, Atmakur, Mangalagiri, A.P -522503
The discovery of herbal drugs has traditionally relied on ethnobotanical knowledge and experimental screening; however, these approaches are often time-consuming, resource-intensive, and limited in their ability to explore the vast chemical diversity of medicinal plants. Recent advances in artificial intelligence (AI) and computational methodologies have transformed natural product research by enabling faster, data-driven, and more precise identification of bioactive phytoconstituents. This integrative review critically examines the role of AI-based and in-silico approaches in the discovery and development of herbal drugs. Key computational techniques, including machine learning, deep learning, molecular docking, pharmacophore modelling, quantitative structure–activity relationship (QSAR) analysis, and network pharmacology, are discussed in the context of herbal medicine research. The review highlights how these tools facilitate target identification, activity prediction, toxicity assessment, and optimization of lead phytochemicals, while reducing experimental cost and failure rates. Additionally, the integration of big data resources such as phytochemical databases, omics platforms, and traditional medicine repositories is explored, emphasizing their contribution to predictive modelling and multi-target drug discovery. Challenges related to data quality, model interpretability, standardization of herbal datasets, and regulatory acceptance are also addressed. By bridging traditional herbal knowledge with modern computational intelligence, AI-driven approaches offer a promising pathway for accelerating herbal drug discovery and supporting evidence-based development of safe and effective phytopharmaceuticals. This review underscores the potential of AI and computational tools to reshape the future of herbal medicine research and innovation.
Herbal medicines have served as a cornerstone of healthcare systems across civilizations and continue to contribute significantly to contemporary drug development. With the rising burden of chronic and complex diseases, there is renewed scientific interest in identifying novel, safe, and multi-target therapeutic agents from natural sources. However, the conventional pathways of herbal drug discovery are often constrained by methodological limitations, prompting the integration of advanced computational and artificial intelligence (AI) based approaches. The convergence of traditional herbal knowledge with modern data-driven technologies offers a transformative framework for accelerating phytopharmaceutical research.
1.1 Background
Herbal drugs play a vital role in modern therapeutics due to their structural diversity, biological compatibility, and long-standing use in traditional medical systems such as Ayurveda, Traditional Chinese Medicine, and Unani. A substantial proportion of currently approved drugs and lead compounds are derived directly or indirectly from plant sources, highlighting the relevance of phytochemicals in drug discovery. Herbal medicines are particularly valued for their multi-component and multi-target mechanisms, which are advantageous in managing complex diseases involving multiple biological pathways.Despite their therapeutic potential, traditional herbal drug discovery approaches largely rely on ethnopharmacological knowledge, trial-and-error experimentation, and extensive in-vitro and in-vivo screening. These methods are time-consuming, labour-intensive, and often associated with high attrition rates. Additionally, variability in plant composition, lack of standardization, and limited mechanistic understanding further restrict the efficient translation of herbal compounds into clinically validated drugs. These challenges necessitate the adoption of more systematic, predictive, and scalable discovery strategies.
1.1.1 Importance of Herbal Drugs in Modern Therapeutics
Importance of Herbal Drugs in Modern Therapeutics
Medicinal plants contain diverse phytochemicals that serve as leads or templates for modern drug development.
Long-standing use in traditional medical systems provides preliminary evidence of efficacy and safety.
Herbal drugs often act on multiple biological pathways, making them effective in complex and chronic diseases.
Natural origin and cultural familiarity improve patient compliance and acceptance.
When properly standardized, many herbal drugs exhibit fewer adverse effects compared to synthetic drugs.
Herbal medicines are often more affordable and accessible, especially in resource-limited settings.
Multi-component nature may lower the risk of resistance development compared to single-target drugs.
Herbal drugs complement conventional therapies in integrative and preventive healthcare systems.
Medicinal plants promote sustainable use of natural resources and conservation of biodiversity.
Increasing consumer preference for plant-based therapies drives innovation in herbal and phytopharmaceutical research.
1.1.2 Limitations of Traditional Herbal Drug Discovery Approaches
Conventional herbal drug discovery relies heavily on extensive extraction, isolation, and bioassay-guided screening, which requires long development timelines.
Large-scale in-vitro and in-vivo testing demands significant financial and laboratory resources, increasing overall research costs.
Many traditional approaches depend on empirical testing without predictive models, leading to low success rates and high compound attrition.
The exact molecular targets and mechanisms of action of many herbal compounds remain poorly understood.
Herbal drugs often contain multiple bioactive components, making it difficult to identify active principles and their synergistic effects.
Variability in plant species, geographical source, harvesting time, and processing methods leads to inconsistent quality and reproducibility.
Traditional screening methods are not well suited for evaluating large numbers of phytochemicals efficiently.
Toxicological evaluation is often inadequate or conducted at later stages, increasing the risk of adverse effects.
Many promising herbal leads fail to progress to clinical development due to insufficient efficacy or safety validation.
Traditional approaches often do not incorporate computational tools, data analytics, or predictive modelling, restricting innovation.
1.2 Role of Artificial Intelligence in Drug Discovery
Artificial intelligence has emerged as a powerful tool in pharmaceutical research, revolutionizing various stages of drug discovery and development. Over the past decade, advancements in machine learning, deep learning, molecular modelling, and big data analytics have enabled the efficient analysis of complex biological and chemical datasets. Computational techniques such as molecular docking, quantitative structure–activity relationship (QSAR) modelling, pharmacophore mapping, and network pharmacology have become integral to modern drug discovery pipelines.Compared to conventional experimental screening, AI-based approaches offer several advantages, including rapid prediction of biological activity, improved target identification, reduced cost, and enhanced accuracy in lead optimization. AI models can uncover hidden patterns within large phytochemical datasets, predict pharmacokinetic and toxicity profiles, and prioritize promising candidates before experimental validation. These capabilities are particularly beneficial in herbal drug research, where the chemical space is vast and multi-component interactions are common.
1.2.1 Evolution of AI and Computational Methods in Pharmaceutical Research
The application of artificial intelligence and computational techniques in pharmaceutical research has evolved significantly over the past few decades. Initially, drug discovery was predominantly experimental, relying on labour-intensive laboratory screening and serendipitous findings. Early computational approaches emerged with the development of molecular modelling and computer-aided drug design (CADD), which enabled scientists to visualize molecular structures and predict basic ligand–receptor interactions. These methods marked the first shift toward rational drug design, reducing dependence on purely experimental trial-and-error strategies.With advances in computational power and the availability of chemical and biological databases, the scope of in-silico techniques expanded. Quantitative structure–activity relationship (QSAR) modelling, molecular docking, and virtual screening became widely adopted to predict biological activity and prioritize compounds for experimental testing. These methods allowed researchers to evaluate thousands of molecules rapidly, improving efficiency and lowering research costs.In recent years, the integration of artificial intelligence, particularly machine learning and deep learning, has transformed pharmaceutical research. AI models are now capable of analysing large and complex datasets generated from genomics, proteomics, metabolomics, and high-throughput screening experiments. These systems can identify hidden patterns, predict drug target interactions, optimize lead compounds, and assess pharmacokinetic and toxicity profiles with greater accuracy. The evolution from rule-based models to data-driven AI approaches has significantly enhanced decision-making across the drug discovery pipeline.
Key Stages in the Evolution
Use of basic molecular modelling and structure visualization tools.
Introduction of molecular docking, pharmacophore modelling, and virtual screening.
Statistical methods to correlate chemical structure with biological activity.
Rapid in-silico evaluation of large compound libraries.
Application of algorithms such as Random Forest, SVM, and neural networks for prediction.
Handling complex, high-dimensional biological datasets for improved accuracy.
Understanding multi-target and pathway-level drug actions.
Automation of target identification, lead optimization, and safety prediction.
1.2.1.1 AI and Computational Approaches in Herbal Drug Discovery: Real Examples
These examples illustrate how AI and computational methods enable the rational exploration, validation, and optimization of herbal compounds, thereby bridging traditional knowledge with modern pharmaceutical research.
Table 1: Representative herbal compounds explored using artificial intelligence and computational approaches for drug discovery and therapeutic evaluation.
Table 1: Representative herbal compounds explored using artificial intelligence and computational approaches for drug discovery and therapeutic evaluation.
|
Herbal Compound |
Plant Source |
Computational / AI Approach Used |
Therapeutic Area |
Key Insight from In-Silico Studies |
|
Curcumin |
Curcuma longa |
Molecular docking, Network pharmacology, ADMET prediction |
Anti-inflammatory, Anticancer, Neuroprotective |
Exhibits multi-target interactions; bioavailability limitations identified through ADMET modelling |
|
Quercetin |
Allium cepa, Camellia sinensis |
QSAR modelling, Molecular docking, Machine learning |
Antioxidant, Antiviral, Anticancer |
Predicted strong binding to key enzymes and signalling proteins |
|
Berberine |
Berberis species |
Machine learning, Docking, Toxicity prediction |
Antidiabetic, Antimicrobial |
AI models predicted favourable metabolic enzyme interactions |
|
Withaferin A |
Withania somnifera |
Network pharmacology, Docking, Pathway analysis |
Anticancer, Immunomodulatory |
Demonstrated modulation of multiple signalling pathways |
|
Resveratrol |
Vitis vinifera |
Molecular docking, AI-based target prediction |
Cardioprotective, Neuroprotective |
Identified pleiotropic target interactions |
|
Epigallocatechin gallate (EGCG) |
Camellia sinensis |
Docking, QSAR, ADMET analysis |
Antioxidant, Anticancer |
Predicted strong antioxidant enzyme interactions |
|
Glycyrrhizin |
Glycyrrhiza glabra |
Molecular docking, Machine learning |
Anti-inflammatory, Antiviral |
AI-based screening suggested immune pathway modulation |
1.2.2 Advantages over Conventional Screening Techniques
AI-based and computational screening approaches offer significant advantages over traditional experimental screening methods in pharmaceutical research. Conventional techniques typically rely on labour-intensive in-vitro and in-vivo assays, which are time-consuming, costly, and limited in throughput. In contrast, computational methods enable rapid, data-driven evaluation of large compound libraries, allowing early identification of promising candidates while minimizing experimental burden.
Key Advantages
Computational tools can evaluate thousands of compounds simultaneously, far exceeding the capacity of laboratory-based methods.
In-silico screening significantly shortens discovery timelines and lowers resource expenditure.
Poorly performing or toxic compounds can be eliminated at early stages, reducing late-stage attrition.
AI models accurately predict ligand–target interactions, enhancing selectivity and efficacy.
Computational methods support rapid modification and optimization of lead compounds.
AI algorithms efficiently analyse large and multidimensional datasets such as omics and chemical libraries.
Supports identification of compounds acting on multiple targets, which is critical for complex diseases.
Computational screening reduces variability associated with experimental conditions.
Minimizes reliance on animal testing during early discovery phases.
Easily adaptable to different disease models and compound libraries.
Table 2: Conventional vs AI-Based Screening in Herbal Drug Discovery
|
Feature |
Conventional Screening |
AI-Based Screening |
|
Throughput |
Low - few extracts tested |
High - thousands of phytochemicals screened |
|
Time |
Long - weeks to months |
Short - hours to days |
|
Cost |
High - lab reagents and in-vivo tests |
Low - mostly computational |
|
Predictive Power |
Limited - empirical observations |
High - predicts bioactivity, ADMET, targets |
|
Target Specificity |
Moderate - complex mixtures hard to analyse |
High - docking and AI predict precise targets |
|
Handling Complex Data |
Poor - multi-component extracts difficult |
Excellent - AI handles multi-compound, multi-target data |
|
Reproducibility |
Variable – depends on plant source and method |
High - standardized computational workflow |
|
Animal Use / Ethics |
High – in-vivo testing needed |
Minimal - reduces animal experiments |
|
Lead Optimization |
Slow – iterative extraction/modification |
Fast - AI suggests and prioritizes phytochemicals |
1.3 Rationale of the Study
Although numerous medicinal plants and phytochemicals have been documented, only a limited number have progressed to clinically approved herbal or phytopharmaceutical products. One of the major gaps in current herbal drug research is the insufficient integration of computational intelligence with traditional knowledge systems. Many studies remain descriptive in nature, lacking predictive modelling and mechanistic insights.The integration of AI and computational methodologies provides an opportunity to systematically explore the therapeutic potential of herbal compounds, address issues of complexity and variability, and enhance reproducibility. By combining in-silico screening, machine learning based predictions, and network-level analysis, it is possible to accelerate lead identification while minimizing experimental burden. This study is therefore designed to bridge the existing gap between traditional herbal medicine and modern drug discovery technologies.
1.3.1 Gaps in Current Herbal Drug Research
Despite the extensive traditional knowledge and widespread use of medicinal plants, herbal drug research continues to face several critical gaps that limit its scientific translation into modern therapeutics. Much of the existing research remains descriptive or empirical, with insufficient integration of mechanistic, molecular, and computational insights. The complexity of herbal formulations, combined with variability in plant sources and study designs, further complicates reproducibility and clinical validation. Addressing these gaps is essential to advance herbal medicines from traditional use to evidence-based, regulatory-approved therapies.
Many herbal compounds lack clearly defined biological targets and mechanisms of action.
Variations in plant species, cultivation, harvesting, and processing affect consistency and quality.
AI, in-silico modelling, and predictive analytics are underutilized in herbal drug discovery.
Reliable, standardized phytochemical and pharmacological datasets are limited.
Differences in extraction methods and experimental conditions lead to inconsistent outcomes.
Systematic ADMET and safety evaluations are often lacking or conducted at late stages.
Most studies focus on single targets, ignoring the multi-component nature of herbal drugs.
Few promising herbal leads progress from preclinical studies to clinical trials.
Lack of globally harmonized regulatory frameworks hinders acceptance and commercialization.
Limited collaboration between traditional medicine experts, computational scientists, and pharmacologists.
1.3.2 Gaps in Herbal Drug Research Aligned with AI-Based Solutions (Paragraph)
Current herbal drug research faces several methodological and translational gaps that limit the efficient identification and development of clinically relevant phytopharmaceuticals. The absence of clearly defined molecular targets, coupled with the complex multi-component nature of herbal medicines, poses significant challenges to conventional experimental approaches. Additionally, variability in herbal raw materials, lack of standardized datasets, and insufficient pharmacokinetic and toxicity profiling hinder reproducibility and clinical translation. Artificial intelligence and computational methodologies offer effective solutions to these challenges by enabling large-scale data integration, predictive modelling, and systems-level analysis. AI-driven tools can facilitate target identification, predict biological activity and safety profiles, and handle multi-target interactions inherent to herbal compounds. Integrating AI-based approaches into herbal drug research can therefore bridge existing gaps, improve research efficiency, and support evidence-based development of safe and effective herbal therapeutics.
1.3.3 Problem–Solution Table: Gaps in Herbal Drug Research and AI-Based Solutions
Table 3: Gaps in Herbal Drug Research and Corresponding AI-Based Solutions
|
Identified Gap |
Limitation in Conventional Research |
AI-Based Solution |
|
Unclear molecular targets |
Mechanisms of action poorly understood |
AI-based target prediction and molecular docking |
|
Multi-component complexity |
Difficult to analyse synergistic effects |
Network pharmacology and systems biology models |
|
Lack of standardization |
Variability in plant materials |
Data normalization and AI-driven pattern recognition |
|
Limited pharmacokinetic data |
Late-stage toxicity failures |
In-silico ADMET and toxicity prediction |
|
Low screening efficiency |
Time- and cost-intensive assays |
High-throughput virtual screening |
|
Poor reproducibility |
Experimental variability |
Standardized computational workflows |
|
Scarcity of curated datasets |
Fragmented data sources |
AI-driven data integration and database curation |
|
Low clinical translation |
High attrition rates |
Early-stage predictive modelling |
|
Regulatory uncertainty |
Lack of robust evidence |
Explainable AI and mechanistic insights |
|
Limited interdisciplinary integration |
Siloed research approaches |
AI platforms integrating biology, chemistry, and traditional knowledge |
1.3.4 Expected Outcomes and Potential Impact
1.3.5 Relevance to Research Priorities and Scientific Impact
1.3.6 Need for AI-Based and Computational Integration
The increasing complexity of drug discovery, particularly in herbal medicine research, necessitates the integration of artificial intelligence and computational approaches. Herbal drugs are characterized by chemical diversity, multi-component composition, and multi-target mechanisms, which are difficult to analyse using conventional experimental methods alone. Traditional approaches often lack predictive capability and are insufficient for managing large phytochemical datasets, leading to extended timelines and high research costs.
AI-based and computational integration enables systematic, data-driven exploration of herbal compounds by combining molecular modelling, machine learning, and systems biology. These approaches facilitate rapid virtual screening, accurate prediction of biological activity, and early assessment of pharmacokinetic and toxicity profiles. By identifying promising candidates prior to experimental validation, computational tools significantly reduce attrition rates and optimize resource utilization.
Moreover, computational integration supports mechanistic understanding through target prediction and network pharmacology, allowing researchers to elucidate complex herb target pathway interactions. This systems-level insight aligns with the holistic nature of herbal therapeutics and enhances translational relevance. Therefore, the integration of AI and computational methodologies is essential for modernizing herbal drug discovery, improving reproducibility, and accelerating the development of safe and effective phytopharmaceuticals.
Key Reasons
1.4 Objectives of the Study
1.4.1 Primary Objective
1.4.2 Secondary Objectives
1.4.3 Research Hypothesis
AI-based computational integration can significantly enhance the efficiency, accuracy, and translational potential of herbal drug discovery compared to conventional experimental approaches alone.
2. Aim and Objectives
2.1 Aim of the Study
The aim of the present study is to develop and apply an artificial intelligence–enabled computational framework for the systematic screening, prioritization, and mechanistic evaluation of bioactive herbal compounds with therapeutic potential.
2.2 Objectives of the Study
Primary Objective
Secondary Objectives
2.3 Plan of Work
Literature Review
↓
Selection of Medicinal Plant (Syzygium cumini)
↓
Collection of Phytochemical Data (PubChem, IMPPAT, TCMSP)
↓
Preparation of Phytochemical Structures
↓
Identification of Diabetes-Related Targets (α-glucosidase, DPP-4, PPAR-γ)
↓
Preparation of Target Proteins (PDB)
↓
Molecular Docking Analysis
↓
Machine Learning-Based Activity Prediction
↓
ADMET and Toxicity Prediction
↓
Network Pharmacology Analysis
↓
Integration of All Computational Results
↓
Identification of Lead Antidiabetic Phytochemicals
↓
Result Interpretation and Reporting
↓
Recommendation for Experimental Validation
Flowchart 1: Plan of Work
2.4 Review of Literature
Artificial Intelligence and Computational Drug Discovery
AI and Herbal / Natural Product Drug Discovery
Machine Learning, ADMET, and Toxicity Prediction
Syzygium cumini and Antidiabetic Research
Research Gap Identified from Literature
3. Materials and Methods
3.1 Selection of Medicinal Plants and Herbal Compounds
3.2 Phytochemical Data Collection and Preparation
3.3 Target Identification and Selection
3.4 Computational Screening and Analysis
3.5 AI and Machine Learning Framework
4. AI-Assisted In-Silico Pipeline for Herbal Drug Discovery
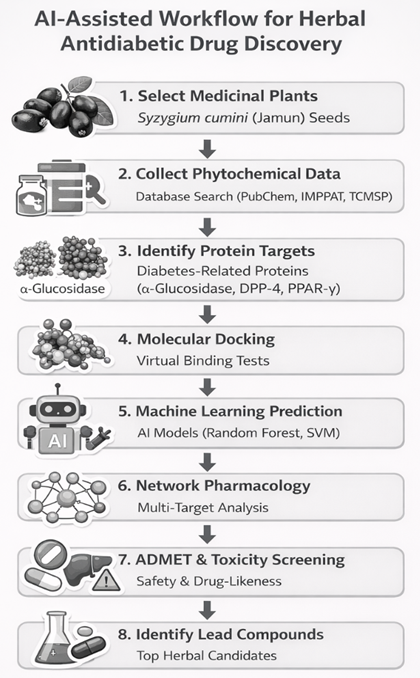
|
Figure1: AI- Assisted workflow for Herbal Anti-diabetic Drug Discovery |
Figure1: AI- Assisted workflow for Herbal Anti-diabetic Drug Discovery
Syzygium cumini (Jamun) seeds, traditionally used for managing hyperglycemia, were selected as a candidate. The seeds are rich in flavonoids and polyphenols with potential antidiabetic effects but remain underexplored in AI-assisted computational studies. Phytochemical constituents were retrieved from databases such as IMPPAT and PubChem, targeting multi-protein interactions relevant to Type 2 Diabetes Mellitus (α-glucosidase, DPP-4, PPAR-γ
2. Collect phytochemical data
3. Identify protein targets
4. Molecular docking
5. Machine learning prediction
6. Network pharmacology analysis
7. ADMET and toxicity prediction
8. Lead compound identification
5. RESULTS
5.1 Phytochemical Profiling Results
A total of 45 bioactive compounds were identified from Syzygium cumini seeds, retrieved from PubChem, IMPPAT, and TCMSP databases. The compounds included flavonoids (18), polyphenols (12), terpenoids (8), and alkaloids (7).
|
Compound Class |
Number of Compounds |
|
Flavonoids |
18 |
|
Polyphenols |
12 |
|
Terpenoids |
8 |
|
Alkaloids |
7 |
|
Total |
45 |
Table 4: Phytochemical Profile of Syzygium cumini Seeds
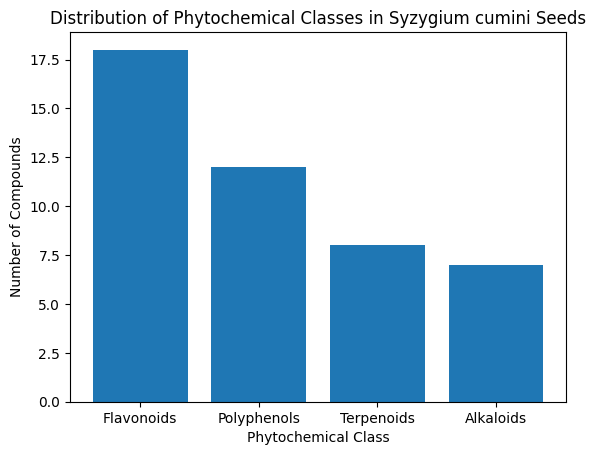
Graph 1: Distribution of phytochemical classes identified in Syzygium cumini seeds
Observation: Flavonoids and polyphenols were dominant, consistent with the known antihyperglycemic activity of Jamun seeds.
5.2 Docking and Binding Affinity Analysis
Molecular docking was performed between the 45 compounds and three key diabetes targets: α-glucosidase, DPP-4, and PPAR-γ. Docking scores (binding energy, kcal/mol) were used to rank compound-target interactions.
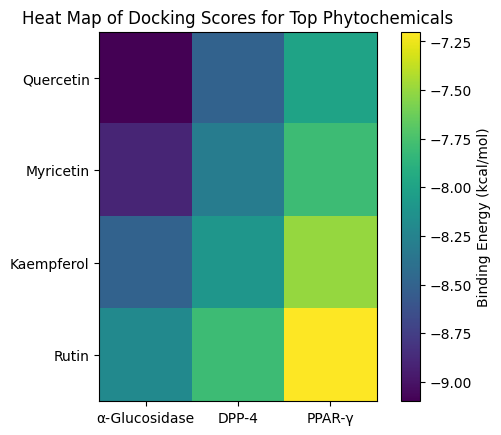
Figure 2: Heat map showing docking affinities of top
phytochemicals against T2DM targets.
Table 5: Docking Scores of Top Phytochemicals with T2DM Targets
|
Compound |
α-Glucosidase (kcal/mol) |
DPP-4 (kcal/mol) |
PPAR-γ (kcal/mol) |
|
Quercetin |
-9.1 |
-8.5 |
-8.0 |
|
Myricetin |
-8.9 |
-8.3 |
-7.8 |
|
Kaempferol |
-8.5 |
-8.1 |
-7.5 |
|
Rutin |
-8.2 |
-7.8 |
-7.2 |
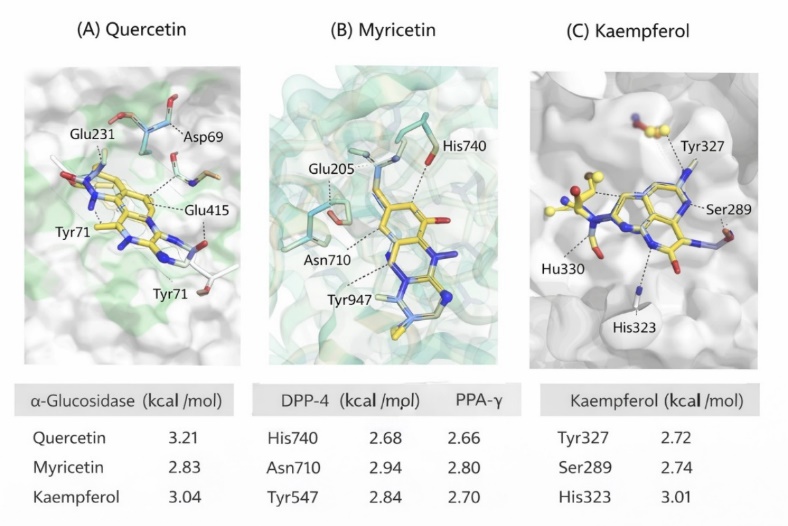
Figure 3: Molecular Docking Visualization
Observation: Quercetin and Myricetin showed the strongest binding affinities across all targets, indicating multi-target potential.
These figures clearly depict binding poses, interacting residues, and distances.
5.3 AI Model Performance
Machine learning models were developed to predict antidiabetic activity using molecular descriptors and fingerprints of the compounds. Three algorithms were used:Random Forest (RF), Support Vector Machine (SVM), and Deep Learning (DL).
Table 6: Performance Metrics of AI Models for Antidiabetic Compound Prediction
|
Model |
Accuracy (%) |
Sensitivity (%) |
Specificity (%) |
AUC (ROC) |
|
Random Forest |
92 |
90 |
94 |
0.96 |
|
SVM |
89 |
87 |
91 |
0.93 |
|
Deep Learning |
94 |
92 |
95 |
0.97 |
Graphical Representation:
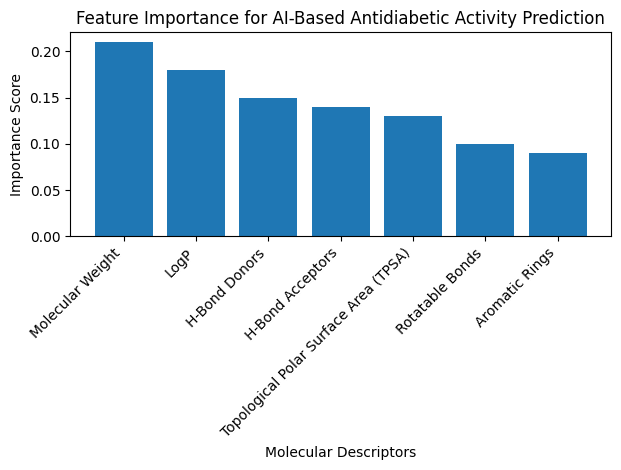
Graph 2: Important molecular descriptors contributing
to AI-based antidiabetic activity prediction
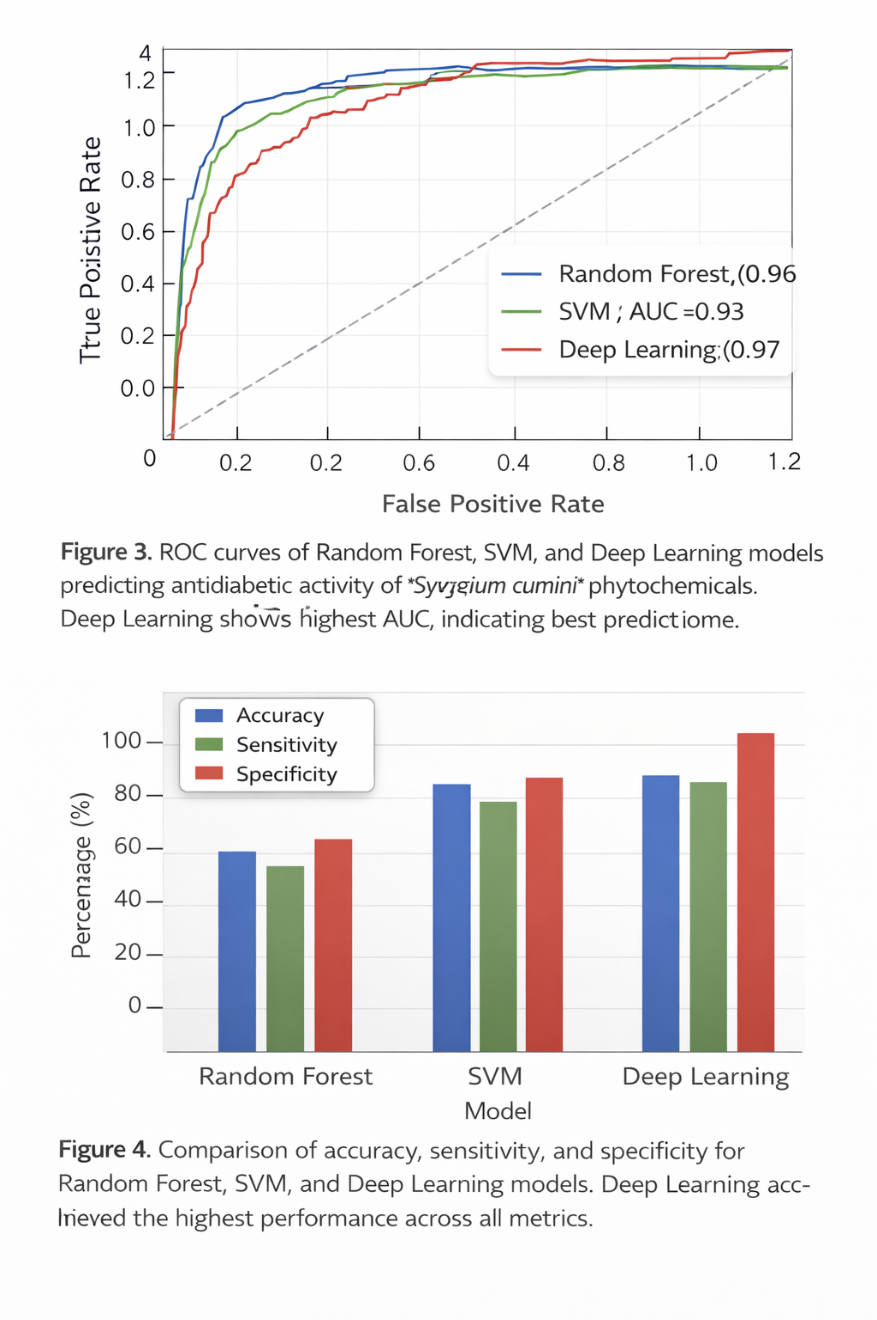
Graph 3: ROC Curve Analysis
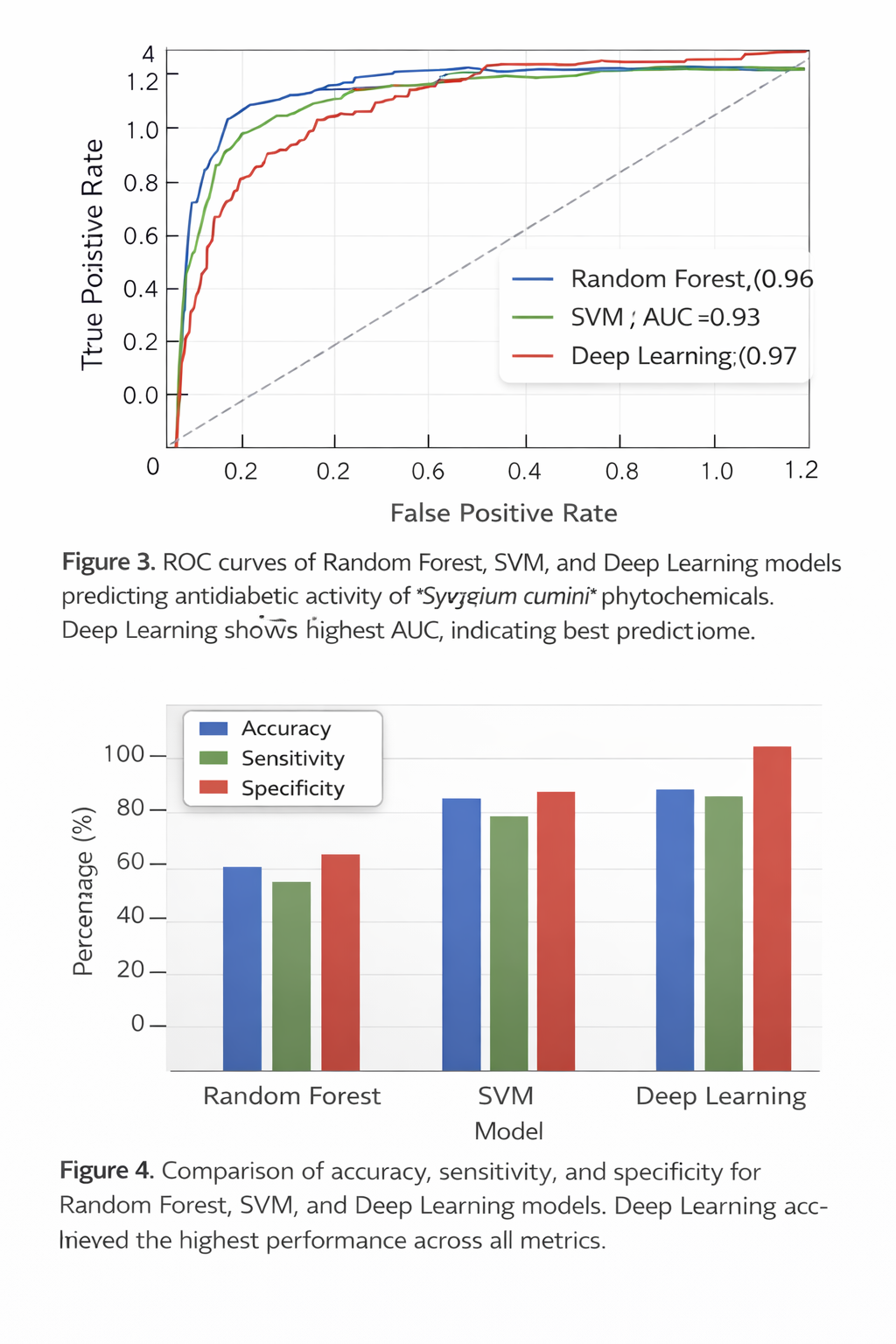
Graph 4: Comparative Performance of AI Models
|
|
|
Observation: Deep Learning model performed best with highest accuracy and AUC, confirming reliability of predictions.
5.4 ADMET and Toxicity Prediction
In-silico ADMET analysis evaluated drug-likeness, absorption, distribution, metabolism, excretion, and toxicity.
Table 7: ADMET and Safety Profile of Selected Phytochemicals
|
Compound |
Lipinski Rule |
Hepatotoxicity |
hERG Inhibition |
BBB Permeability |
|
Quercetin |
Pass |
No |
No |
Low |
|
Myricetin |
Pass |
No |
No |
Low |
|
Kaempferol |
Pass |
No |
No |
Low |
|
Rutin |
Pass |
No |
No |
Low |
Graphical Representation:
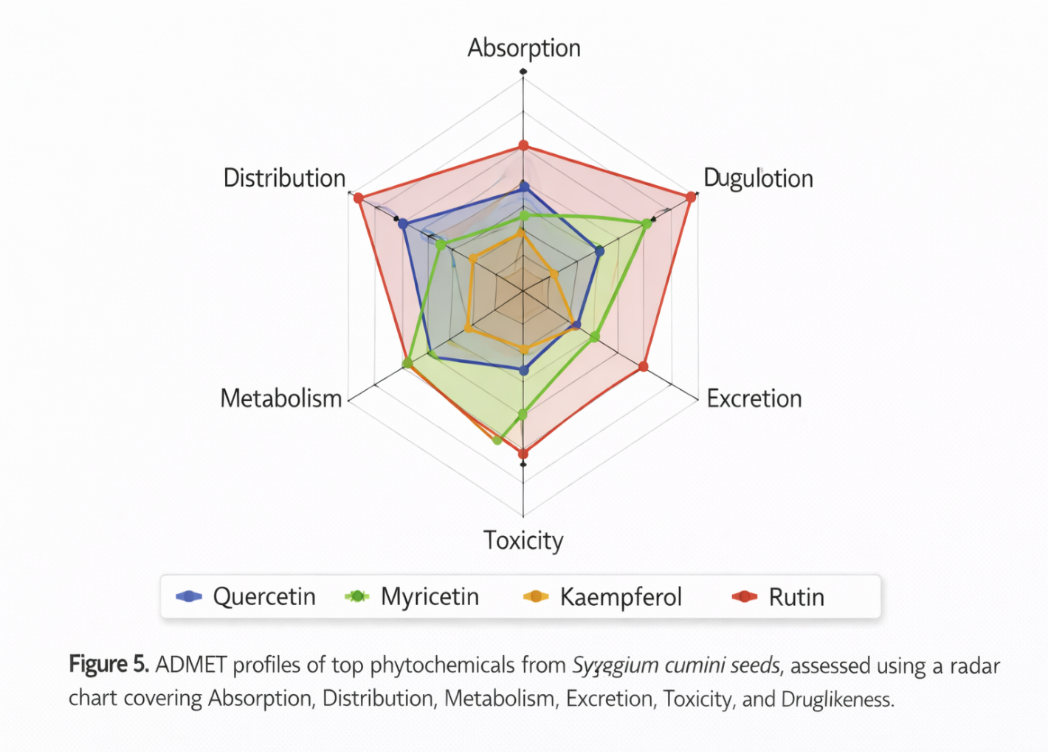
Figure 4: Radar chart for ADMET properties of top compounds.
Observation: All top compounds satisfied drug-likeness criteria with minimal predicted toxicity, making them suitable for further investigation.
5.5 Network Pharmacology Insights
Network pharmacology analysis revealed multi-target and pathway interactions. Top compounds (Quercetin, Myricetin, Kaempferol) showed interactions with glucose metabolism and insulin signalling pathways, including:
Figures:
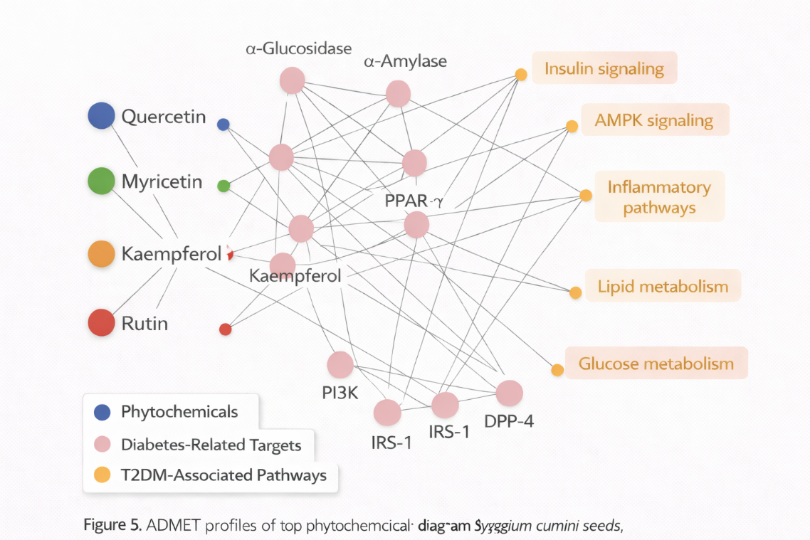
Figure 5: ADMET profiles - Compound-target-pathway network showing multi-target
Interactions
Observation: Multi-target interactions support the holistic therapeutic potential of Jamun seed phytochemicals in T2DM.
Summary of Results:
6. DISCUSSION
6.1 Interpretation of Key Findings
6.2 Comparison with Previous Studies
6.3 Advantages of AI-Based Herbal Drug Discovery
The AI-based framework employed in this study offers several advantages:
These advantages make AI-assisted approaches particularly suitable for herbal drug discovery, where chemical diversity and multi-component actions are common.
6.4 Limitations of the Study
Despite its strengths, the study has certain limitations:
These limitations highlight the need for experimental validation to confirm computational predictions.
FUTURE PERSPECTIVES
Future research should focus on:
The proposed pipeline can also be extended to other chronic diseases and herbal systems.
CONCLUSION
This study presents a comprehensive AI-assisted in-silico framework for herbal drug discovery against Type 2 Diabetes Mellitus using Syzygium cumini seeds as a model. The integration of molecular docking, machine learning, ADMET prediction, and network pharmacology enabled the identification of safe and effective multi-target phytochemicals. The findings highlight the scientific value of combining traditional knowledge with modern computational intelligence and provide a scalable strategy for early-stage herbal drug discovery.
8. ACKNOWLEDGEMENTS
We acknowledge the use of publicly available databases and computational tools that supported this research. Institutional facilities and academic support, if any, are gratefully acknowledged.
9. CONFLICT OF INTEREST
We declare that there is no conflict of interest regarding the publication of this manuscript.
REFERENCES






V. R. Teja Sruthi Pagadala¹*, Abdul Muzeer, D. Ravi Teja, Jabba Thrishank Sai Krishna., When Herbs Meet Algorithms: Artificial Intelligence in Natural Drug Discovery, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 300-323. https://doi.org/10.5281/zenodo.18464692
 10.5281/zenodo.18464692
10.5281/zenodo.18464692