The nicotinamide adenine dinucleotide (NAD?) salvage pathway plays a central role in cellular metabolism, redox balance, DNA repair, and gene regulation. In this article, we have comprehensively discussed the impact of pharmacogenetic variants in key NAD? salvage pathway enzymes including NAMPT, NAPRT1, NMNAT1–3, NRK1/2, PARP1, CD38, and SIRT1 on drug response, efficacy, and toxicity. The NAD? salvage pathway maintains cellular redox homeostasis, DNA repair, and metabolic regulation; thus, genetic variations in these enzymes can profoundly alter pharmacological outcomes. We have explored the physiological and biological importance of NAD?, its interaction with drug metabolism, and the comparative analysis of NAD? biosynthesis routes. Furthermore, the article highlights how these variants influence drug efficacy and toxicity, emphasizing the role of companion diagnostics and pharmacogenetic testing in guiding personalized therapy. By integrating clinical and molecular data, this review underscores the potential of NAD? pathway genotyping to improve treatment precision, reduce adverse reactions, and enhance patient-specific therapeutic success.
Keywords
NAD? salvage pathway, pharmacogenomics, NAMPT, NAPRT, PARP1, SIRT1, drug response, toxicity, personalized medicine
Introduction
×
The concept of personalized medicine seeks to tailor medical treatment to the individual characteristics of each patient. Among the key factors enabling this approach are pharmacogenomics and pharmacogenetics, which study how genetic variations influence drug efficacy and toxicity. One of the emerging areas of interest is the NAD? (nicotinamide adenine dinucleotide) salvage pathway, a major route for NAD? biosynthesis in mammalian cells. NAD? is a critical cofactor involved in redox reactions, cellular signalling, DNA repair, and aging. Perturbations in NAD? levels are linked to cancer, neurodegeneration, metabolic diseases, and immune dysfunction. Notably, pharmacogenetic variants in the genes coding for enzymes of the NAD? salvage pathway has been found to influence drug metabolism and therapeutic responses1. This review provides a comprehensive exploration of how genetic polymorphisms in NAD? salvage pathway enzymes modulate drug response and how these insights can be integrated into personalized medicine2.
2. OVERVIEW OF THE NAD? SALVAGE PATHWAY
The NAD? salvage pathway recycles nicotinamide (NAM), the product of NAD?-consuming reactions, back into NAD?. The key enzymes involved include:
NAMPT (Nicotinamide phosphoribosyl transferase): Converts NAM to nicotinamide mononucleotide (NMN).
NMNATs (Nicotinamide mononucleotide adenylyl transferases): Convert NMN to NAD?.
CD38 and PARPs: Consume NAD? in signalling and DNA repair, indirectly influencing the salvage pathway.
This pathway is preferred over de novo NAD? synthesis in most human tissues due to its energy efficiency and tight regulation.
PHYSIOLOGICAL ROLE OF NAD?3
The NAD? salvage pathway restores NAD? from nicotinamide (NAM) and nicotinic acid (NA). Key enzymes include:
NRKs (nicotinamide riboside kinases) and additional peripheral enzymes control tissue- specific precursor usage. These enzymes determine cellular NAD? pools and thereby regulate activity of NAD?-consuming drug-relevant enzymes (PARPs, sirtuins, CD38), so genetic variation that changes their expression or activity can alter drug effects.
KNOWN PHARMACOGENETIC / FUNCTIONAL VARIANTS4
NAMPT: Multiple SNPs and haplotypes have been reported. Specific variants (e.g., rs1319501 and rs4730153) have been associated with altered circulating NAMPT (visfatin) levels and with differences in clinical phenotypes (metabolic syndrome traits, preeclampsia biomarkers) and drug responses (e.g., antihypertensive response in one study). Functional consequences include altered NAMPT expression or secretion.
NAPRT1: Shows substantial genetic diversity, somatic or germline variation (or epigenetic silencing) in tumors is frequent and can determine whether a cell depends on NAMPT or NAPRT for NAD? synthesis. This diversity is clinically important because NAPRT-deficient tumors are often sensitized to NAMPT inhibitors or to combinations with DNA-damaging agents.
NMNATs: Rare loss-of-function variants in NMNAT1 have been linked to inherited retinal disease (LCA) and neurodegeneration, indicating tissue vulnerability when NAD biosynthesis is impaired. While not classical “pharmacogenetic” variants yet, NMNAT variation can change cellular NAD homeostasis and may modulate responses to drugs that stress NAD-consuming systems (e.g., PARP activators).
Other NAD-related genes: Broader surveys have identified many rare coding variants across the NAD metabolic network that are predicted to be functional — these are candidate pharmacogenetic variants that merit follow-up.
MECHANISTIC LINKS — HOW VARIANTS CHANGE DRUG RESPONSE4
Altering basal NAD? levels — variants that lower NAMPT/NMNAT/NAPRT activity reduce NAD? pools, which can:
Increase sensitivity to energetic stress and to drugs that further deplete NAD? (NAMPT inhibitors, some chemotherapies).
Reduce activity of sirtuins and PARPs, changing DNA repair capacity and epigenetic regulation (modifying chemo or radiotherapy response).
Shifting precursor dependence — NAPRT1 loss or low expression means cells rely on NAMPT; such tumors may be selectively killed by NAMPT inhibitors (therapeutic window). Conversely, tumors with intact NAPRT can resist NAMPT inhibition or be rescued by NA supplementation.
Modulating non-metabolic roles of enzymes — NAMPT has extracellular signalling roles (visfatin) and influences inflammation and vascular factors; genetic variants altering secretion could influence pharmacodynamics of cardiovascular or anti-inflammatory drugs.
Interaction with PARP inhibitors — PARP inhibitors create reliance on NAD?/DNA repair resources. Tumors with defective NAD biosynthesis (e.g., low NAPRT or NAMPT variants) may be more or less sensitive depending on context — prompting combination strategies (NAMPT inhibitors + PARP inhibitors) in specific genotypes.
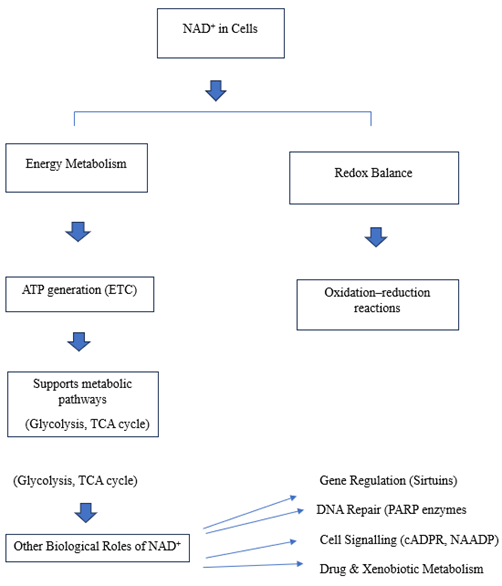
FIGURE 1: FLOWCHART: OVERVIEW OF NAD? FUNCTIONS14
CLINICAL & TRANSLATIONAL IMPLICATIONS
Predictive biomarkers: Germline or somatic genotyping (NAMPT, NAPRT1, NMNATs) may predict which patients will benefit from NAD-targeting therapies (NAMPT inhibitors, NAD precursor supplementation) or from PARP/chemo combinations. Tumor NAPRT1 status is already explored as a predictive marker for NAMPT inhibitor sensitivity.
Drug selection / dosing: Patients with germline variants that reduce systemic NAD may be more susceptible to toxicity from NAD-depleting drugs (require dose adjustments); conversely, variants that increase NAD synthesis might blunt therapeutic efficacy of NAMPT inhibitors.
Companion diagnostics: Measuring tumor NAPRT1 expression (IHC / methylation / sequencing) or genotyping NAMPT SNPs could serve as companion diagnostics for trials of NAMPT inhibitors or NAD-modulating therapies.
Repurposing / combination therapies: Combining NAMPT inhibitors with PARP inhibitors or standard chemotherapies may produce synthetic lethality in NAD-vulnerable genotypes — but requires stratification by genotype and careful toxicity monitoring.
STUDY DESIGNS TO DEMONSTRATE PHARMACOGENETIC EFFECTS4
Population genetics + phenotype association: large cohorts (multi-ethnic) genotyped for candidate SNPs (NAMPT rs1319501, rs4730153, NAPRT variants) correlated with drug response endpoints (response rate, toxicity, survival). Include both germline and tumor somatic status.
Functional assays: express variants in cell lines (or use CRISPR knock-ins) and measure NAD? levels, PARP/sirtuin activity, and drug sensitivity (NAMPT inhibitors, PARP inhibitors, chemotherapeutics). Rescue experiments with NA/NR/NMN help prove causality.
Tumor models: xenograft or PDX models stratified by NAPRT1 expression/genotype to test NAMPT inhibitors ± PARP inhibitors. Monitor systemic NAD and toxicity.
Clinical trial design suggestions: biomarker-driven phase I/II trials where patients are pre-stratified by tumor NAPRT1 status or by NAMPT genotype; include pharmacodynamic readouts (tumor NAD, Arylation levels) and safety biomarkers.
GAPS, CHALLENGES & FUTURE DIRECTIONS
Paucity of definitive pharmacogenetic associations: While mechanistic rationale and preclinical data are strong, large, replicated clinical studies directly linking specific germline variants to drug response are still limited. More population-scale genotyping plus drug response data is needed.
Complex tissue specificity: NAD metabolism is tissue- and precursor-dependent; a variant that matters in one tissue (e.g., retina or hematopoietic cells) may be irrelevant in another. Functional readouts must therefore be tissue-matched.
Rare variants matter: Many functional variants are rare; standard GWAS may miss them. Deep sequencing and functional annotation pipelines are required to capture rare but high-impact pharmacogenetic alleles
INTERACTION WITH DRUG METABOLISM
The NAD? (Nicotinamide Adenine Dinucleotide) molecule plays a central role in many chemical reactions that help the body metabolize or break down drugs. It acts as a coenzyme, meaning it helps enzymes work properly during oxidation–reduction (redox) reactions.
When NAD? levels are normal, liver enzymes—especially those responsible for drug metabolism—can function efficiently. However, when NAD? levels decrease (because of enzyme defects or genetic variations), drug metabolism can become slower or imbalanced, leading to either drug accumulation (toxicity) or reduced therapeutic effect.5
Role of NAD? in Drug-Metabolizing Reactions
(a) Phase I Reactions – Functionalization
Phase I reactions mainly involve oxidation, reduction, or hydrolysis, carried out by enzymes such as cytochrome P450s (CYPs), alcohol dehydrogenase (ADH), and aldehyde dehydrogenase (ALDH).
These enzymes often need NAD? or its phosphorylated form NADP? to transfer electrons during oxidation.
For example:
Alcohol dehydrogenase uses NAD? to convert ethanol → acetaldehyde.
Aldehyde dehydrogenase again uses NAD? to convert acetaldehyde → acetic acid.
Without enough NAD?, these steps slow down, causing toxic buildup (like acetaldehyde in alcohol intolerance).6
(b) Phase II Reactions – Conjugation
NAD? also indirectly supports energy balance required for conjugation reactions (e.g., glucuronidation, sulfation, acetylation).
When NAD? is depleted, the energy supply for these reactions drops, so drug excretion becomes slower.
HOW THE NAD? SALVAGE PATHWAY AFFECTS THIS PROCESS
The salvage pathway regenerates NAD? from precursors such as nicotinamide (vitamin?).7
TABLE 1. REPRESENTS MAIN ENZYMES INVOLVED ARE 7:
Enzyme
Function
If Gene Variant Occurs
NAMPT
Converts nicotinamide → NMN (rate-limiting step)
Reduced NAD? production → slower redox reactions and metabolism
NAPRT1
Converts nicotinic acid → NaMN (alternate pathway)
Cells rely only on NAMPT; vulnerable to NAD? depletion
NMNAT1-3
Convert NMN → NAD?
If mutated, NAD? pool decreases → less support for detoxifying enzymes
INTERACTION WITH CYTOCHROME P450 SYSTEM
The CYP450 system (responsible for metabolizing ~75% of all drugs) depends on NADPH, which is made from NADP? (derived from NAD?).
So, if the NAD? salvage pathway is weak, less NADPH is available to drive CYP450 activity.9
This leads to:
Reduced metabolism rate
Higher plasma concentration of drugs
Greater risk of side effects or toxicity.
Conversely, overactive NAD? synthesis (in some individuals) may cause faster metabolism, reducing the drug’s therapeutic effect.10
5. Drug-Induced Effects on NAD? Levels
Some drugs also consume or deplete NAD? during their metabolism:
Isoniazid (anti-TB drug) and valproic acid reduce NAD? levels in the liver.
Ethanol metabolism heavily uses NAD?, leading to redox imbalance.
In people with weak NAD?-producing enzymes (due to genetic variation), such drugs can cause hepatotoxicity or enhanced side effects because NAD? gets exhausted faster than it can be replenished.
6. Personalized Medicine Perspective
Pharmacogenetic testing can help identify individuals with NAD? salvage pathway variants, such as:
NAMPT (rs1319501, rs4730153)
NAPRT1 (loss or silencing variants)
Adjust drug doses for better safety and efficacy.
Avoid drugs that excessively deplete NAD?.
Recommend NAD? precursors (e.g., nicotinamide riboside or nicotinic acid) to maintain normal metabolism.11
BIOLOGICAL IMPORTANCE OF NAD?
NAD? is a vital coenzyme found in all living cells
It acts like a “cellular battery”, helping enzymes transfer energy and electrons during metabolic reactions.12
It continuously cycles between two forms:
NAD? (oxidized form) – accepts electrons
NADH (reduced form) – donates electrons
This continuous conversion is essential for:
Energy production
DNA repair
Gene regulation
Cell signalling
Aging and immunity 13
TABLE 2. REPRESENTS BIOLOGICAL ROLES 15
Function
Description
Outcome / Importance
1.Energy Production
NAD? accepts electrons during glycolysis and TCA cycle; NADH donates them in the electron transport chain.
Generates ATP for all cellular activities.
2.Redox Reactions
Maintains the balance between oxidation and reduction.
Keeps metabolism and cell homeostasis stable.
3. DNA Repair
Serves as substrate for PARP enzymes to repair DNA damage.
Preserves genomic integrity and prevents mutations.
4.Gene Regulation (Sirtuins)
Activates NAD?-dependent sirtuin enzymes.
Controls aging, inflammation, and stress response.
5. Cell Signalling
Precursor of cADPR and NAADP, which regulate Ca²? signalling.
Controls muscle contraction, immunity, and nerve signals.
6. Immune and Inflammatory Control
Regulates energy use in immune cells, reduces oxidative stress.
Supports immune defense and reduces chronic inflammation.
Prevents neurodegenerative diseases and supports brain health.
TABLE 3: REPRESENTS COMPARISON OF NAD? BIOSYNTHESIS ROUTES 16
Pathway
Starting Material
Main Enzyme Involved
Tissue Distribution
Advantages
Limitations
De novo Pathway
Tryptophan
TDO/IDO, ACMSD, QPRT
Liver, kidney
Operates even without niacin intake
Slow; energy-intensive; can form neurotoxic intermediates
Preiss–Handler Pathway
Nicotinic acid
NAPRT, NMNAT
Most tissues
Efficient conversion responsive to niacin therapy
Depends on dietary nicotinic acid
Salvage Pathway
Nicotinamide /Nicotinamide riboside
NAMPT, NMNAT, NRK
All tissues
Fast, energy-efficient; major source in cells
Impaired by mutations in NAMPT or NAD?-consuming enzymes
PHARMACOGENETIC VARIANTS IN NAD? SALVAGE ENZYMES 17
NAMPT Variants
NAMPT is a rate-limiting enzyme in NAD? salvage. Variants such as rs61330082 (T>C) and rs9770242 have been associated with metabolic disorders, altered immune responses, and cancer susceptibility.
Drug impact: NAMPT inhibitors (e.g., FK866) are being developed as anticancer agents. Patients with certain NAMPT polymorphisms may exhibit variable sensitivity or resistance.
Clinical implication: A variant causing reduced NAMPT activity could predispose individuals to enhanced drug toxicity or decreased therapeutic efficacy due to compromised NAD? regeneration.
NMNAT Variants
There are three isoforms: NMNAT1 (nuclear), NMNAT2 (cytosolic), and NMNAT3 (mitochondrial).
Variants in NMNAT2 have been linked to neurodegenerative conditions and may affect drug response to neuroprotective agents or NAD? boosters like nicotinamide riboside.
NMNAT1 polymorphisms (e.g., rs2302559) are under study for associations with retinal degeneration and could influence drug response in ocular therapies.
CD38 and Other NAD?-Consuming Enzymes
CD38 modulates immune cell function and inflammation through NAD? consumption. Variants like rs3796863 and rs6449182 have been linked to insulin resistance, inflammation, and autoimmune disease.
Therapeutic context: CD38 inhibitors are explored for cancer immunotherapy. Genetic variants might predict which patients benefit most or suffer adverse immune effects.
TABLE 4. KEY NAD? SALVAGE PATHWAY ENZYMES AND THEIR GENETIC VARIANTS 18
Variants influence NAD? production and metabolic drug responses, particularly in lipid metabolism.
NRK2
Nicotinamide Riboside Kinase 2 – Converts NR → NMN in muscle tissues
rs17009016
SNP associated with altered energy metabolism; affects muscle response to NAD?-boosting supplements.
PARP1
Poly(ADP-ribose) Polymerase 1 – Consumes NAD? during DNA repair
rs1136410 (Val762Ala)
Decreased enzyme activity; affects DNA repair and response to PARP inhibitors used in cancer therapy.
CD38
NAD? Glycohydrolase – Regulates NAD? degradation
rs6449182, rs1800561
Polymorphisms influence NAD? degradation rate; affect immune response and metabolism-related drug efficacy.
SIRT1
Sirtuin 1 – NAD?-dependent deacetylase involved in gene regulation
rs7069102, rs7895833
Variants linked to altered deacetylase activity and varied response to anti-aging or metabolic drugs.
CLINICAL SIGNIFICANCE OF THESE VARIANTS 18
NAMPT (Nicotinamide Phosphoribosyl transferase)
Variants such as rs61330082 and rs2302559 in the NAMPT gene alter the enzyme’s expression and activity, leading to fluctuations in intracellular NAD? levels. Clinically, these changes are associated with metabolic disorders, including type 2 diabetes, obesity, and inflammation.
In oncology, altered NAMPT activity can affect a patient’s response to NAMPT inhibitors like FK866 and CHS-828, which are used in cancer treatment. Therefore, identifying NAMPT variants helps tailor drug dosage and predict therapeutic outcomes.
Mutations such as rs10498357 and rs6641 in NMNAT1 reduce enzyme stability and NAD? production in the nucleus. This has been linked to Leber congenital amaurosis, a retinal degeneration disease, and increased vulnerability to oxidative stress.
Clinically, patients with these variants may show a reduced response to drugs that depend on oxidative balance or antioxidant mechanisms, such as neuroprotective agents.
3. NMNAT2 and NMNAT3 19
Variants in NMNAT2 (e.g., rs181930065) influence axonal NAD? synthesis, which is essential for neuronal survival. These changes are associated with neurodegenerative diseases like Alzheimer’s and Parkinson’s. Similarly, NMNAT3 variants (e.g., rs28528368) affect mitochondrial NAD? production, potentially leading to energy metabolism disorders. Such mutations can modify the effectiveness of mitochondrial-targeted therapies or NAD?-enhancingsupplements in neuroprotection or cancer management.
4. NAPRT (Nicotinate Phosphoribosyl transferase)
Polymorphisms like rs2066807 and rs975457 in the NAPRT gene decrease the enzyme’s ability to utilize nicotinic acid (niacin).
Clinically, these variants are significant in cancer therapy, where NAPRT-deficient tumors show higher sensitivity to NAMPT inhibitors such as FK866 and GMX1778. This makes NAPRT status a key biomarker for personalized oncology, helping clinicians predict which patients will respond to NAD?-depleting anticancer drugs.
5. NRK1 and NRK2 (Nicotinamide Riboside Kinases)
Variants in NRK1 (NMRK1), such as rs838133 and rs10498745, and in NRK2 (NMRK2), such as rs17009016, affect the conversion of nicotinamide riboside (NR) into NMN, influencing tissue-specific NAD? regeneration.
These changes have been linked to altered lipid metabolism, muscle energy disorders, and variable responses to NAD? precursor supplements (NR, NMN). In clinical settings, such variants help determine who will benefit most from NAD?-boosting therapies for metabolic or muscular diseases.
6. PARP1 (Poly ADP-Ribose Polymerase 1)
The rs1136410 (Val762Ala) variant in PARP1 decreases its catalytic activity in DNA repair. This is clinically relevant in cancer and cardiovascular diseases. Reduced PARP1 activity increases susceptibility to DNA damage but also enhances sensitivity to PARP inhibitors such as Olaparib and niraparib, used in cancer treatment. Hence, PARP1 genotyping is crucial for optimizing targeted cancer therapy.
7. CD38 (NAD? Glycohydrolase)
Variants like rs6449182 and rs1800561 in CD38 lead to increased NAD? degradation and altered immune cell signalling. These genetic differences are associated with insulin resistance, chronic inflammation, and age-related metabolic decline.
Clinically, they affect the response to CD38 inhibitors (e.g., daratumumab, used in multiple myeloma) and influence the effectiveness of NAD? supplementation in metabolic or aging therapies.
8. SIRT1 (Sirtuin 1)
Variants such as rs7069102 and rs7895833 in the SIRT1 gene affect its NAD?-dependent deacetylase activity, influencing gene regulation and metabolic pathways. These polymorphisms are linked with type 2 diabetes, cardiovascular disorders, and longevity. Clinically, SIRT1 variants can determine a patient’s response to SIRT1 activators like resveratrol or SRT2104, which are used to improve metabolism and delay aging processes.
4. CLINICAL IMPLICATIONS FOR DRUG RESPONSE 20
4.1 Cancer Therapy
NAD? metabolism is crucial in supporting cancer cell survival. Inhibitors of NAMPT deplete NAD? levels, inducing tumor cell death. However, pharmacogenetic variations can:
Modify tumor sensitivity to NAMPT inhibitors.
Alter toxicity profiles in patients, especially affecting hematopoietic or GI tissues.
Influence efficacy of DNA-damaging agents (e.g., PARP inhibitors), as NAD? levels affect DNA repair capacity.
4.2 Anti-inflammatory and Autoimmune Therapies 21
Given NAD?'s role in immunomodulation, variations in its pathway enzymes influence responses to corticosteroids, TNF inhibitors, and newer biologics.
Example: CD38 overexpression is associated with lupus and rheumatoid arthritis; its variants could impact responsiveness to immunosuppressive drugs.
4.3 Neurological and Psychiatric Disorders
In neurodegeneration (e.g., Alzheimer’s, Parkinson’s), NAD? depletion is a pathological hallmark. NAD? precursors are being tested as therapeutic agents.
Genetic variants in NMNAT2 or NAMPT may affect outcomes of NAD?-boosting strategies.
Personalized NAD? therapy could improve cognitive function and delay neurodegeneration in genetically predisposed individuals.
4.4 Metabolic Disorders 22
Obesity, diabetes, and NAFLD have been associated with NAD? dysregulation. Pharmacogenetic profiling could help:
Predict metformin or SGLT2 inhibitor responsiveness.
Determine benefits from NAD? precursors or sirtuin activators.
Drug efficacy: NAPRT-deficient tumors respond better to NAMPT inhibitors (FK866, GMX1778).
Drug toxicity: Normal cells may experience enhanced NAD? depletion–related toxicity.
Clinical example: FK866 treatment in pancreatic cancer; dosing must be carefully managed 27.
5. NRK1 & NRK2 (Nicotinamide Riboside Kinases)
Effect: Impaired NAD? regeneration from nicotinamide riboside.
Drug efficacy: Reduced response to NAD?-boosting supplements (NR, NMN).
Drug toxicity: Increased risk of muscle fatigue or metabolic side effects during supplementation or NAD?-dependent therapies.
Clinical example: Nicotinamide riboside supplementation in patients with metabolic syndrome may show variable benefits. 28
6. PARP1 (Poly ADP-Ribose Polymerase 1)
Effect: Decreased DNA repair capacity.
Drug efficacy:PARP inhibitors (e.g., Olaparib, niraparib) may work more effectively in some variants.
Drug toxicity: Higher risk of genotoxicity, bone marrow suppression, and mucositis with DNA-damaging agents.
Clinical example: Olaparib in BRCA-mutated ovarian or breast cancer; toxicity risk higher in PARP1 Val762Ala carriers. 29
7. CD38 (NAD? Glycohydrolase)
Effect: Increased NAD? degradation.
Drug efficacy: Reduced efficacy of NAD? supplementation or CD38-targeted drugs.
Drug toxicity: Enhanced immune-related adverse effects and cytotoxicity from NAD?-lowering drugs.
Clinical example: Daratumumab therapy in multiple myeloma; NAD?-dependent side effects may increase in certain CD38 variants. 30
8. SIRT1 (Sirtuin 1)
Effect: Altered deacetylase activity.
Drug efficacy: Reduced response to SIRT1 activators (resveratrol, SRT2104) and some metabolic therapies.
Drug toxicity: Higher susceptibility to metabolic drug toxicity or oxidative stress–related damage.
Clinical example: Resveratrol supplementation in type 2 diabetes patients may be less effective with SIRT1 polymorphisms. 31
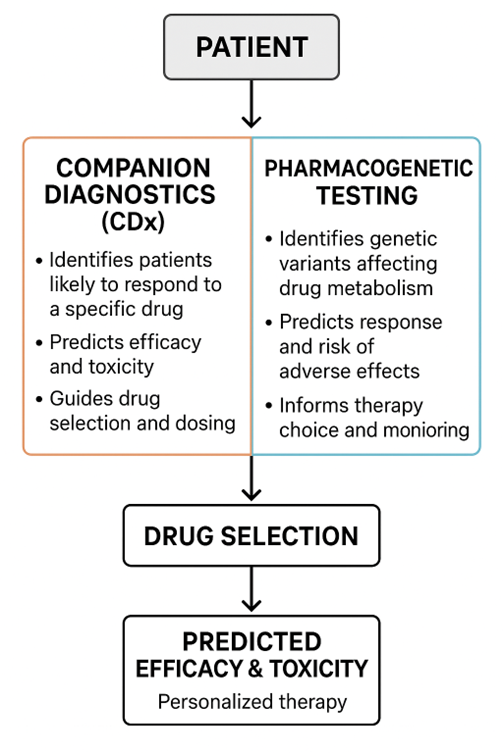
COMPANION DIAGNOSTICS AND PHARMACOGENETIC TESTING
Companion Diagnostics (CDx) 32
Companion diagnostics are specialized tests or assays designed to identify patients who are most likely to benefit from a particular therapeutic drug, or who may be at increased risk of adverse effects.
These diagnostics provide crucial molecular or genetic information that guides clinicians in choosing the right drug, the right dose, and the right patient population.
Purpose
Predict Drug Response: Identify patients who will respond positively to a targeted therapy.
Minimize Toxicity: Prevent severe adverse effects by identifying patients with genetic susceptibilities.
Optimize Treatment: Determine drug selection and dosing to maximize therapeutic efficacy.
Mechanism of Action
CDx relies on detecting biomarkers, protein expression levels, or specific genetic variants that influence how a drug interacts with its target or is metabolized in the body.
In the context of NAD? metabolism, this often involves testing the expression or activity of salvage pathway enzymes (like NAMPT, NAPRT, or PARP1) to understand how a patient’s cells will respond to NAD?-modulating drugs.33
Examples in NAD? Salvage Pathway–Targeted Therapy
NAMPT and NAPRT Testing:
NAMPT inhibitors (e.g., FK866, CHS-828) are designed to deplete NAD? levels in tumor cells.
CDx testing identifies tumors that are NAPRT-deficient, as these tumors are more susceptible to NAD? depletion, while normal tissues expressing NAPRT are protected.
This helps clinicians select patients who will benefit most and avoid severe toxicity in those with normal NAPRT expression. 34
PARP inhibitors (e.g., Olaparib, niraparib) are more effective in patients with BRCA mutations or reduced PARP1 activity.
CDx testing allows precise identification of patients likely to respond to therapy and those at risk for DNA damage–related toxicity.
CLINICAL SIGNIFICANCE
CDx supports personalized medicine by:
Guiding drug choice and therapy strategy.
Reducing trial-and-error in drug selection.
Enhancing treatment outcomes, particularly in cancer therapy, metabolic disorders, and neurodegenerative diseases. 35
Pharmacogenetic Testing
Pharmacogenetic testing examines individual genetic variations that affect drug metabolism, pharmacodynamics, and pharmacokinetics.
Unlike CDx, which is often linked to a specific drug-target interaction, pharmacogenetic testing considers multiple genes that influence how a drug is absorbed, distributed, metabolized, and excreted, as well as how it affects cellular pathways. 36
Purpose
Predict Efficacy: Determine whether a patient is likely to respond to a particular drug.
Assess Toxicity Risk: Identify patients susceptible to adverse drug reactions due to enzyme deficiencies or altered drug metabolism.
Enable Dose Optimization: Adjust therapeutic doses according to genetic makeup to achieve maximal benefit with minimal toxicity. 37
Lower intracellular NAD?; influence response to NAMPT inhibitors (FK866).
Patients with these variants may experience enhanced cytotoxicity in normal tissues, requiring dose adjustment.
NMNAT1/2/3 Variants:
Alter NAD? production in the nucleus, axons, or mitochondria.
Affect neuroprotective drugs, mitochondrial therapies, and susceptibility to chemotherapy-induced neuropathy.38
NRK1/NRK2 Variants:
Influence conversion of nicotinamide riboside to NMN.
Determine effectiveness of NAD? precursor supplements in metabolic or age-related therapies.
CD38 Variants:
Increase NAD? degradation.
Affect efficacy of CD38-targeted drugs (e.g., daratumumab) and NAD? supplementation therapies.
SIRT1 Variants:
Alter NAD?-dependent deacetylase activity.
Influence response to SIRT1 activators like resveratrol or SRT2104 and metabolic drugs.39
PARP1 Variants:
Reduced DNA repair activity; alter efficacy of PARP inhibitors (Olaparib, niraparib).
Increase risk of genotoxicity when treated with DNA-damaging chemotherapy.
3. CLINICAL APPLICATIONS WITH DRUG EXAMPLES
FK866 (NAMPT inhibitor):
Pharmacogenetic testing for NAMPT/NAPRT informs patient selection and dose to maximize efficacy and minimize toxicity.
Olaparib (PARP inhibitor):
Testing for PARP1 or BRCA mutations guides therapy and predicts toxicity risk.
Nicotinamide Riboside/NMN Supplements:
NRK1/NRK2 testing predicts who will benefit most from NAD?-boosting therapy.
Resveratrol or SIRT1 Activators:40
SIRT1 variants determine drug response and metabolic side effect risk.
Advantages of Pharmacogenetic Testing
Facilitates precision dosing and drug selection.
Reduces adverse drug reactions and toxicity.
Enhances therapeutic outcomes across oncology, neuroprotection, and metabolic therapies
Figure 2: FRAMEWORK FOR PERSONALIZED NAD?-TARGETED INTERVENTIONS.41
5.1 INTEGRATION INTO PERSONALIZED MEDICINE
Genotyping for Clinical Decisions
Routine genotyping for NAD? salvage enzyme variants is not yet standard. However, as pharmacogenomic panels expand, including genes like NAMPT, NMNAT1/2/3, CD38, and PARP1 could provide:
Better prediction of adverse drug reactions.
Identification of responders to NAD? modulators or salvage pathway inhibitors.
Stratification of patients in clinical trials involving NAD?-targeted therapies 42
Development of Biomarkers
NAD? levels in tissues or blood, along with genotypic data, may serve as dual biomarkers to guide therapy. For instance: 43
Low NAD? with a loss-of-function NAMPT variant could justify lower drug doses.
High CD38 expression combined with activating variants might indicate a need for immune modulation.
Potential for Polypharmacy Optimization
In patients taking multiple drugs—especially in oncology or geriatrics—NAD? pathway variants could inform drug interactions and help avoid depletion-related toxicity.44
CURRENT CHALLENGES AND FUTURE DIRECTIONS
Lack of Large-Scale Genetic Data
Many studies on NAD? pathway variants are based on small cohorts or preclinical models. More large-scale, multi-ethnic GWAS are needed.
Limited Pharmacogenomic Trials
Few clinical trials stratify patients based on NAD? salvage pathway genotypes. This gap delays the translation of pharmacogenetic knowledge into practice. 45
Complex Interactions with Diet and Environment
NAD? levels are influenced by exercise, caloric intake, and vitamin B3 status. Integrating environmental and lifestyle data is essential to fully understand drug-genome interactions.
Cost and Accessibility
Widespread genetic testing and NAD? level monitoring remain costly and are not uniformly available across healthcare systems. 46
CONCLUSION
Pharmacogenetic variations in NAD? salvage pathway enzymes significantly influence drug metabolism, efficacy, and safety. Understanding these genetic differences enables clinicians to predict therapeutic response, minimize toxicity, and tailor treatment strategies. Incorporating companion diagnostics and pharmacogenetic testing into clinical practice will advance precision medicine by ensuring that NAD?-targeted therapies are both effective and safe for each individual.
REFERENCES
Ying, W. (2008). NAD? and NADH in cellular functions and cell death. Frontiers in Bioscience, 13, 3129–3148.
Houtkooper, R. H., Canto, C., Wanders, R. J., & Auwerx, J. (2010). The secret life of NAD?: An old metabolite controlling new metabolic signaling pathways. Endocrine Reviews, 31(2), 194–223.
Garten, A., Petzold, S., Körner, A., Imai, S., & Kiess, W. (2009). Nampt: Linking NAD biology, metabolism, and cancer. Trends in Endocrinology & Metabolism, 20(3), 130–138.
Canto, C., Menzies, K. J., & Auwerx, J. (2015). NAD? metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus. Cell Metabolism, 22(1), 31–53.
Zhang, T., & Kraus, W. L. (2010). SIRT1-dependent regulation of chromatin and transcription: Linking NAD? metabolism and signaling to the control of cellular functions. Biochemicals et Biophysical Acta, 1804(8), 1666–1675.
Chiarugi, A., Dölle, C., Felici, R., & Ziegler, M. (2012). The NAD metabolome—A key determinant of cancer cell biology. Nature Reviews Cancer, 12(11), 741–752.
Rajman, L., Chwalek, K., & Sinclair, D. A. (2018). Therapeutic potential of NAD?-boosting molecules: The in vivo evidence. Cell Metabolism, 27(3), 529–547.
Yoshino, J., Baur, J. A., & Imai, S. (2018). NAD? intermediates: The biology and therapeutic potential of NAD? precursors. Nature Reviews Molecular Cell Biology, 19(9), 585–606.
Bogan, K. L., & Brenner, C. (2008). Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD? precursor vitamins in human nutrition. Annual Review of Nutrition, 28, 115–130.
Hasmann, M., & Schema Inda, I. (2003). FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyl transferase. Cancer Research, 63(21), 7436–7442.
Canto, C., Houtkooper, R. H., Pirinen, E., et al. (2012). The NAD? precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet–induced obesity. Cell Metabolism, 15(6), 838–847.
Frederick, D. W., Loro, E., Liu, L., et al. (2016). Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metabolism, 24(2), 269–282.
Bajrami, I., Kigozi, A., Van Wever Wijk, A., et al. (2012). Synthetic lethality between PARP and NAMPT inhibition in triple-negative breast cancer. Nature Communications, 3, 1–10.
Watson, M., Roulston, A., Belec, L., et al. (2009). Pharmacological inhibition of NAMPT as a therapeutic strategy in hematologic malignancies. Molecular Cancer Therapeutics, 8(9), 2555–2564.
Tarragó, M. G., Crisol, B., de Los Ríos, C., et al. (2018). A role for NAD? in the regulation of immune function and inflammation. Frontiers in Immunology, 9, 1–15.
Katsuya, E., Mottis, A., Zietak, M., et al. (2018). De novo NAD? synthesis enhances mitochondrial function and improves health span in mice. Nature Metabolism, 1(7), 609–619.
romero, M. F., & Brenner, C. (2017). Role of NMNAT enzymes in neural protection and degeneration. Journal of Neurochemistry, 142(5), 678–692.
Pollak, N., Dölle, C., & Ziegler, M. (2007). The power to reduce: pyridine nucleotides—small molecules with a multitude of functions. Biochemical Journal, 402(2), 205–218.
Chakraborty, S., & Ahuja, N. (2019). CD38 and age-related NAD? decline: Therapeutic perspectives. Trends in Pharmacological Sciences, 40(1), 1–12.
Belenky, P., Bogan, K. L., & Brenner, C. (2007). NAD? metabolism in health and disease. Trends in Biochemical Sciences, 32(1), 12–19.
Gardell, L. R., Jeong, S. M., & Licht, J. (2019). Clinical development of NAD?-modulating therapies: challenges and opportunities. Clinical Pharmacology & Therapeutics, 106(4), 744–756.
Uddin, M. E., Kabir, N., & Chandrani, P. (2020). Pharmacogenomics and NAD metabolism: implications for personalized therapy. Pharmacogenomics Journal, 20(5), 647–660.
Pradère, J. P., & Sadler, A. J. (2017). NMNAT and neuroprotection: molecular pathways and therapeutic strategies. Neuroscience Letters, 652, 72–81.
Wang, Y., & Tissenbaum, H. A. (2018). NAD? and aging: mechanisms and intervention strategies. Cell Research, 28(9), 891–905.
Vyas, S., Chesarone-Cataldo, M., Todorova, T., et al. (2013). A systematic analysis of PARP1 function in DNA repair and chromatin regulation. Molecular Cell, 49(4), 612–623.
Fang, E. F., Lautrup, S., Hou, Y., et al. (2017). NAD? in aging: molecular mechanisms and translational implications. Trends in Molecular Medicine, 23(10), 899–916.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461(7267), 1071–1078.
Sánchez-Ramírez, B., & Cruces-Solís, H. (2020). NAPRT1 loss and cancer sensitivity to NAD? depletion: preclinical evidence and therapeutic prospects. Cancer Research, 80(14), 2890–2900.
Rajman, L., & Sinclair, D. (2016). The role of NAD? in neuronal health and neurodegeneration. Annual Review of Neuroscience, 39, 91–117.
Grabowska, I., & Roberts, P. J. (2019). Metabolic reprogramming and NAD? in cancer progression. Cancer Metabolism, 7(1), 1–14.
Yamaguchi, S., & Imai, S. (2011). NAD? and sirtuins in metabolic disease and longevity. Clinical Science, 120(4), 143–162.
Jackson, S. P. (2016). PARP inhibitors: clinical development and mechanisms of resistance. British Journal of Cancer, 114(7), 895–903.
Pollak, N. (2013). The role of NAD? metabolome in cancer therapy. Nature Reviews Cancer, 13(10), 713–720.
Grozio, A., Katsyuba, E., & Brenner, C. (2019). Therapeutic potential of nicotinamide riboside and related NAD? precursors. Annual Review of Pharmacology and Toxicology, 59, 171–192.
Srinivasan, S., & Guillemin, G. J. (2018). Kynurenine pathway metabolism and NAD?: intersections in neurodegeneration. Neurochemistry International, 120, 1–11.
Zhou, Q., & Sun, X. (2019). NAMPT genetic variation and metabolic disease risk: population studies and functional analyses. Human Genetics, 138(6), 607–620.
Berger, F., Sauer, U., & Ziegler, M. (2004). Regulation of NAD? synthesis: the interplay between nicotinamide phosphoribosyl transferase and nicotinic acid phosphoribosyl transferase. Journal of Biological Chemistry, 279(21), 21757–21763.
Smirnova, N. A., & Cox, B. (2017). CD38 inhibitors in clinical development: implications for cancer and age-related diseases. Drug Discovery Today, 22(11), 1680–1691.
Morevati, M., & D’Mello, S. R. (2015). NMNATs in axon survival and neuroprotection. Neurobiology of Disease, 78, 46–57.
Martens, C. R., Denman, B. A., Mazzo, M. R., et al. (2018). Chronic nicotinamide riboside supplementation is well tolerated and elevates NAD? in healthy adults: a randomized, placebo-controlled trial. Cell Metabolism, 28(4), 738–748.
Ullmann, U., & Krüger, A. (2014). Genetic variants in PARP1 and therapy responses: implications for oncology. Pharmacogenomics, 15(9), 1205–1218.
Ratajczak, J., Joffraud, M., Trammell, S. A., et al. (2016). NRK1 controls nicotinamide riboside metabolism in mammalian cells. Nature Communications, 7, 1–13.
Sacco, M., Russo, R., Locatelli, F., et al. (2019). CD38 genetic polymorphisms and metabolic disease: population studies and functional assays. Metabolism, 94, 1–10.
Li, J., & Wang, D. (2017). SIRT1 polymorphisms and metabolic disease risk: a meta-analysis. PLoS ONE, 12(5), e0176920.
Camacho-Pereira, J., Tarragó, M. G., Chini, C. C. S., et al. (2016). CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metabolism, 23(6), 1127–1139.
Guarente, L. (2013). Calorie restriction and sirtuins revisited. Genes & Development, 27(19), 2072–2085.
Reference
Ying, W. (2008). NAD? and NADH in cellular functions and cell death. Frontiers in Bioscience, 13, 3129–3148.
Houtkooper, R. H., Canto, C., Wanders, R. J., & Auwerx, J. (2010). The secret life of NAD?: An old metabolite controlling new metabolic signaling pathways. Endocrine Reviews, 31(2), 194–223.
Garten, A., Petzold, S., Körner, A., Imai, S., & Kiess, W. (2009). Nampt: Linking NAD biology, metabolism, and cancer. Trends in Endocrinology & Metabolism, 20(3), 130–138.
Canto, C., Menzies, K. J., & Auwerx, J. (2015). NAD? metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus. Cell Metabolism, 22(1), 31–53.
Zhang, T., & Kraus, W. L. (2010). SIRT1-dependent regulation of chromatin and transcription: Linking NAD? metabolism and signaling to the control of cellular functions. Biochemicals et Biophysical Acta, 1804(8), 1666–1675.
Chiarugi, A., Dölle, C., Felici, R., & Ziegler, M. (2012). The NAD metabolome—A key determinant of cancer cell biology. Nature Reviews Cancer, 12(11), 741–752.
Rajman, L., Chwalek, K., & Sinclair, D. A. (2018). Therapeutic potential of NAD?-boosting molecules: The in vivo evidence. Cell Metabolism, 27(3), 529–547.
Yoshino, J., Baur, J. A., & Imai, S. (2018). NAD? intermediates: The biology and therapeutic potential of NAD? precursors. Nature Reviews Molecular Cell Biology, 19(9), 585–606.
Bogan, K. L., & Brenner, C. (2008). Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD? precursor vitamins in human nutrition. Annual Review of Nutrition, 28, 115–130.
Hasmann, M., & Schema Inda, I. (2003). FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyl transferase. Cancer Research, 63(21), 7436–7442.
Canto, C., Houtkooper, R. H., Pirinen, E., et al. (2012). The NAD? precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet–induced obesity. Cell Metabolism, 15(6), 838–847.
Frederick, D. W., Loro, E., Liu, L., et al. (2016). Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metabolism, 24(2), 269–282.
Bajrami, I., Kigozi, A., Van Wever Wijk, A., et al. (2012). Synthetic lethality between PARP and NAMPT inhibition in triple-negative breast cancer. Nature Communications, 3, 1–10.
Watson, M., Roulston, A., Belec, L., et al. (2009). Pharmacological inhibition of NAMPT as a therapeutic strategy in hematologic malignancies. Molecular Cancer Therapeutics, 8(9), 2555–2564.
Tarragó, M. G., Crisol, B., de Los Ríos, C., et al. (2018). A role for NAD? in the regulation of immune function and inflammation. Frontiers in Immunology, 9, 1–15.
Katsuya, E., Mottis, A., Zietak, M., et al. (2018). De novo NAD? synthesis enhances mitochondrial function and improves health span in mice. Nature Metabolism, 1(7), 609–619.
romero, M. F., & Brenner, C. (2017). Role of NMNAT enzymes in neural protection and degeneration. Journal of Neurochemistry, 142(5), 678–692.
Pollak, N., Dölle, C., & Ziegler, M. (2007). The power to reduce: pyridine nucleotides—small molecules with a multitude of functions. Biochemical Journal, 402(2), 205–218.
Chakraborty, S., & Ahuja, N. (2019). CD38 and age-related NAD? decline: Therapeutic perspectives. Trends in Pharmacological Sciences, 40(1), 1–12.
Belenky, P., Bogan, K. L., & Brenner, C. (2007). NAD? metabolism in health and disease. Trends in Biochemical Sciences, 32(1), 12–19.
Gardell, L. R., Jeong, S. M., & Licht, J. (2019). Clinical development of NAD?-modulating therapies: challenges and opportunities. Clinical Pharmacology & Therapeutics, 106(4), 744–756.
Uddin, M. E., Kabir, N., & Chandrani, P. (2020). Pharmacogenomics and NAD metabolism: implications for personalized therapy. Pharmacogenomics Journal, 20(5), 647–660.
Pradère, J. P., & Sadler, A. J. (2017). NMNAT and neuroprotection: molecular pathways and therapeutic strategies. Neuroscience Letters, 652, 72–81.
Wang, Y., & Tissenbaum, H. A. (2018). NAD? and aging: mechanisms and intervention strategies. Cell Research, 28(9), 891–905.
Vyas, S., Chesarone-Cataldo, M., Todorova, T., et al. (2013). A systematic analysis of PARP1 function in DNA repair and chromatin regulation. Molecular Cell, 49(4), 612–623.
Fang, E. F., Lautrup, S., Hou, Y., et al. (2017). NAD? in aging: molecular mechanisms and translational implications. Trends in Molecular Medicine, 23(10), 899–916.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461(7267), 1071–1078.
Sánchez-Ramírez, B., & Cruces-Solís, H. (2020). NAPRT1 loss and cancer sensitivity to NAD? depletion: preclinical evidence and therapeutic prospects. Cancer Research, 80(14), 2890–2900.
Rajman, L., & Sinclair, D. (2016). The role of NAD? in neuronal health and neurodegeneration. Annual Review of Neuroscience, 39, 91–117.
Grabowska, I., & Roberts, P. J. (2019). Metabolic reprogramming and NAD? in cancer progression. Cancer Metabolism, 7(1), 1–14.
Yamaguchi, S., & Imai, S. (2011). NAD? and sirtuins in metabolic disease and longevity. Clinical Science, 120(4), 143–162.
Jackson, S. P. (2016). PARP inhibitors: clinical development and mechanisms of resistance. British Journal of Cancer, 114(7), 895–903.
Pollak, N. (2013). The role of NAD? metabolome in cancer therapy. Nature Reviews Cancer, 13(10), 713–720.
Grozio, A., Katsyuba, E., & Brenner, C. (2019). Therapeutic potential of nicotinamide riboside and related NAD? precursors. Annual Review of Pharmacology and Toxicology, 59, 171–192.
Srinivasan, S., & Guillemin, G. J. (2018). Kynurenine pathway metabolism and NAD?: intersections in neurodegeneration. Neurochemistry International, 120, 1–11.
Zhou, Q., & Sun, X. (2019). NAMPT genetic variation and metabolic disease risk: population studies and functional analyses. Human Genetics, 138(6), 607–620.
Berger, F., Sauer, U., & Ziegler, M. (2004). Regulation of NAD? synthesis: the interplay between nicotinamide phosphoribosyl transferase and nicotinic acid phosphoribosyl transferase. Journal of Biological Chemistry, 279(21), 21757–21763.
Smirnova, N. A., & Cox, B. (2017). CD38 inhibitors in clinical development: implications for cancer and age-related diseases. Drug Discovery Today, 22(11), 1680–1691.
Morevati, M., & D’Mello, S. R. (2015). NMNATs in axon survival and neuroprotection. Neurobiology of Disease, 78, 46–57.
Martens, C. R., Denman, B. A., Mazzo, M. R., et al. (2018). Chronic nicotinamide riboside supplementation is well tolerated and elevates NAD? in healthy adults: a randomized, placebo-controlled trial. Cell Metabolism, 28(4), 738–748.
Ullmann, U., & Krüger, A. (2014). Genetic variants in PARP1 and therapy responses: implications for oncology. Pharmacogenomics, 15(9), 1205–1218.
Ratajczak, J., Joffraud, M., Trammell, S. A., et al. (2016). NRK1 controls nicotinamide riboside metabolism in mammalian cells. Nature Communications, 7, 1–13.
Sacco, M., Russo, R., Locatelli, F., et al. (2019). CD38 genetic polymorphisms and metabolic disease: population studies and functional assays. Metabolism, 94, 1–10.
Li, J., & Wang, D. (2017). SIRT1 polymorphisms and metabolic disease risk: a meta-analysis. PLoS ONE, 12(5), e0176920.
Camacho-Pereira, J., Tarragó, M. G., Chini, C. C. S., et al. (2016). CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metabolism, 23(6), 1127–1139.
Guarente, L. (2013). Calorie restriction and sirtuins revisited. Genes & Development, 27(19), 2072–2085.
Aditi Tyagi
Corresponding author
Neotech Institute of Pharmacy, Neotech technical Campus Virod, Vadodara, Gujarat 390022
Harsh Parmar
Co-author
Neotech Institute of Pharmacy, Neotech technical Campus Virod, Vadodara, Gujarat 390022
Harsh Parmar, Aditi Tyagi, Impact of Pharmacogenetic Variants in NAD? Salvage Pathway Enzymes on Drug Response: A Personalized Medicine Perspective, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 2028-2045. https://doi.org/10.5281/zenodo.18318084



 10.5281/zenodo.18318084
10.5281/zenodo.18318084