1Department of Chemistry, New Government Polytechnic, Patna (Bihar), India
2Department of Chemistry, Sitamarhi Institute of Technology, Sitamarhi (Bihar), India
3Department of Physics, Sitamarhi Institute of Technology, Sitamarhi (Bihar), India
In the present work, a comprehensive first-principles investigation of the geometrical and electronic properties of chlorpheniramine, a widely used anti-allergic drug, has been carried out using density functional theory (DFT). The optimized molecular geometry of chlorpheniramine was obtained at the B3LYP/6-311++G(d,p) level of theory, and the structural parameters were analyzed to understand the stability and molecular configuration of the compound. The electronic properties were examined through frontier molecular orbital (FMO) analysis, including the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). The HOMO–LUMO energy gap provides valuable insight into the chemical stability, reactivity, and charge transfer characteristics of the molecule. Furthermore, the molecular electrostatic potential (MESP) surface was mapped to identify the electron-rich and electron-deficient regions responsible for possible electrophilic and nucleophilic interactions. The MESP analysis reveals the distribution of electrostatic potential over the molecular surface, highlighting the potential reactive sites within the drug molecule. The combined geometrical optimization, HOMO–LUMO analysis, and MESP mapping provide a detailed understanding of the electronic structure and reactivity of chlorpheniramine. These theoretical findings contribute to a deeper insight into the physicochemical behavior of chlorpheniramine and may be useful for further pharmacological and molecular design studies related to anti-allergic drugs.
Antihistamines represent an important class of pharmaceutical compounds widely used for the treatment of allergic disorders such as rhinitis, urticaria, and conjunctivitis [1-4]. Among these, chlorpheniramine is a well-known first-generation antihistamine that acts as a histamine H? receptor antagonist and is commonly prescribed to relieve symptoms associated with seasonal allergies and the common cold [5]. Due to its significant pharmacological activity and widespread clinical use, understanding the molecular structure and electronic characteristics of chlorpheniramine is important for elucidating its physicochemical behavior, intermolecular interactions, and potential reactivity [6-9].In recent years, computational chemistry has become an essential tool for investigating the structural and electronic properties of biologically active molecules [10-16]. First-principles or ab initio methods based on density functional theory (DFT) provide reliable insights into molecular geometry, charge distribution, frontier molecular orbitals, and electronic transitions without relying on empirical parameters. These theoretical approaches enable detailed analysis of molecular systems at the atomic level and complement experimental techniques such as spectroscopy and crystallographyThe geometrical parameters of a molecule play a crucial role in determining its chemical stability and biological activity [17, 18]. Accurate prediction of bond lengths, bond angles, and molecular conformation helps in understanding the structural features responsible for molecular interactions with biological receptors. Furthermore, the study of electronic properties, including the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), provides valuable information about molecular reactivity, charge transfer processes, and chemical stability. The HOMO–LUMO energy gap is particularly important in determining the kinetic stability and electronic excitation behavior of a molecule.
In addition to structural and electronic characteristics, the optical properties of organic molecules have attracted increasing attention due to their relevance in photochemical processes and molecular interactions with electromagnetic radiation. Parameters such as polarizability, electronic transitions, and absorption characteristics can be effectively evaluated using quantum chemical calculations. Such analyses help in understanding the interaction of molecules with light and may provide insights into their spectroscopic signatures and potential applications in optoelectronic or photochemical systems. Chlorpheniramine contains aromatic rings, heteroatoms, and conjugated systems that may contribute significantly to its electronic distribution and optical response. Despite its pharmacological importance, comprehensive theoretical investigations focusing on its geometrical optimization, electronic structure, and optical behavior remain limited. Therefore, a detailed computational study is necessary to explore these properties and provide deeper insight into its molecular characteristics. In the present work, a first-principles investigation of chlorpheniramine has been carried out to analyze its geometrical structure, electronic properties, and optical characteristics. The molecular geometry was optimized using density functional theory, and the resulting structural parameters were analyzed to understand the stability and conformation of the molecule. Furthermore, frontier molecular orbital analysis, energy gap evaluation, and optical property calculations were performed to elucidate the electronic behavior and photophysical characteristics of chlorpheniramine. The results obtained from this study provide valuable theoretical information that may contribute to a better understanding of the physicochemical and electronic features of this pharmaceutically important compound.
2. Computational details
The ab initio calculations employ a hybrid form of density functional, B3LYP in conjunction with 6-311++G(d, p) basis set for geometry optimization. The B3LYP combines Becke’s three parameter exchange term [19] with the functional devised by Lee-Yang-Parr to treat electron correlation [20, 21]. The Gaussian 09 package [22] is used to perform all computations and visual animation generated by GaussView 5.0 program [23]. The optimized geometry of chlorpheniramine has been shown in Figure 1. The structures of chlorpheniramine was minimized without any constraint in the potential energy surface at B3LYP level, adopting the standard 6-311++G(d,p) basis set.
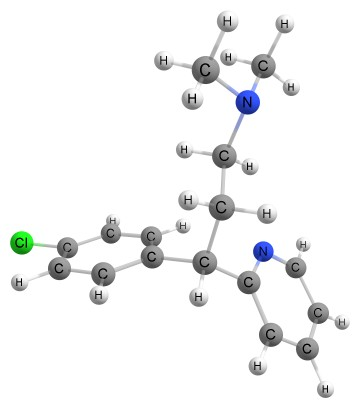
Figure 1. Optimized geometry of chlorpheniramine at B3LYP/6-311++G(d,p) level of theory
RESULT AND DISCUSSION
3.1. Frontier molecular orbital surfaces and MESP plot
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the important electronic parameters associated with the orbital in a compound and the difference between them, results in an HOMO-LUMO energy gap. The HOMO primarily acts as an electron donor and the LUMO largely acts as the electron acceptor. The HOMO and LUMO plot of Chlorpheniramine is displayed in Figure 2. These orbitals find out the way the molecule interacts with other species and their HOMO-LUMO energy gap helps to describe the kinetic stability and chemical reactivity of the molecule [24, 25]. Thus the HOMO-LUMO energy gap is an important parameter measure for stability index of the compound [26] and establishes correlations in various chemical systems [27]. The frontier orbital electron densities on atoms of compound provide a useful means for the characterization of donor-acceptor interactions in details. The HOMO-LUMO energy gap of chlorpheniramine is 4.55 eV. The importance of the MESP (molecular electrostatic potential) lies in the fact that it simultaneously displays the molecular size and shape, as well as positive, negative, and neutral electrostatic potential regions in terms of the electrostatic surface. It is very useful in the investigation of the most probable binding receptor site along with the size and shape of the molecules [28-32] and the MESP plots of chlorpheniramine is displayed in Figure 3. One can see that in MESP, most electropositive i.e. acceptor (the blue part) is located at CH while most electronegative centre, i.e. donor (red part) is located at CH3 atoms. From Figure 3, it is clear that benzene ring possesses a delocalized charge, so can act as a suitable centre of positive charge.
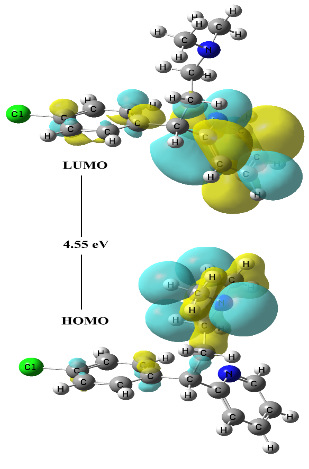
Figure 2. HOMO-LUMO plots of Chlorpheniramine,
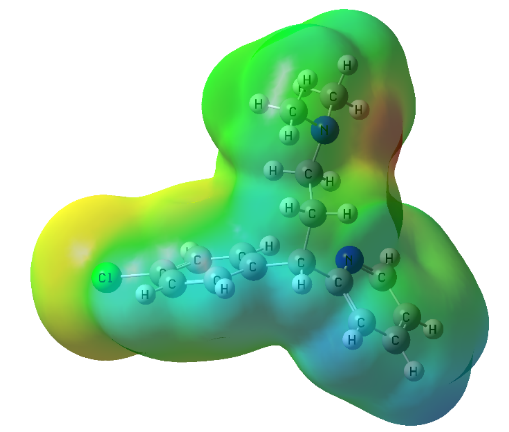
Figure 3. MESP plot of Chlorpheniramine,
3.2. Electronic and thermodynamic parameters
The thermodynamic and electronic parameters of Chlorpheniramine calculated at B3LYP/6-311++G (d, p) level are listed in Table 1. These calculated parameters are used to describe chemical reactivity of molecules. The calculated electronic parameters include electron affinity (A), ionization potential (I), chemical hardness (η) and absolute electro negativity (χ). Ionization potential and electron affinity are calculated as the negative of energy eigen-values of HOMO and LUMO respectively. χ and η can be calculated by using finite-difference approximation [33] as below,
χ = ½(I + A) and η = ½(I – A).
Thermodynamical parameters such as zero point energy (ZPE), thermal energy at room temperature (E), entropy (S), heat capacity (Cv) and enthalpy (H) may be useful in estimating reaction path of the molecules.
Table 1. Electronic and Thermodynamic parameters calculated at B3LYP/6-311++G(d,p) level,

CONCLUSION
In this study, the geometrical and electronic properties of chlorpheniramine were systematically investigated using density functional theory at the B3LYP/6-311++G(d,p) level of theory. The optimized molecular structure revealed a stable geometry with well-defined bond lengths and bond angles, indicating the structural stability of the molecule. The frontier molecular orbital analysis provided valuable insight into the electronic behaviour of the compound. The distribution of the HOMO and LUMO orbitals and the calculated energy gap suggest moderate chemical stability and potential charge transfer within the molecule. Furthermore, the molecular electrostatic potential (MESP) surface analysis was performed to visualize the charge distribution and identify possible reactive regions. The MESP map indicated that the negative potential regions are mainly localized around electronegative atoms, while the positive potential regions are distributed over hydrogen atoms and less electronegative parts of the molecule. These regions are important for understanding possible electrophilic and nucleophilic interactions. Overall, the combined analysis of optimized geometry, HOMO–LUMO orbitals, and MESP surface provides a detailed understanding of the electronic structure, stability, and reactive sites of chlorpheniramine. The results obtained from this theoretical study may serve as a useful reference for further computational and experimental investigations related to the pharmacological properties and molecular interactions of anti-allergic drugs.
REFERENCES



Ram Naresh Singh, Arun Kumar, Ratnesh Kumar, A First Principle Study on Geometrical and Electronic Properties of Chlorpheniramine: An Anti-Allergic Drug, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 3203-3209, https://doi.org/10.5281/zenodo.19229030
 10.5281/zenodo.19229030
10.5281/zenodo.19229030
