
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Mederametla Anjamma Mastan Rao College of Pharmacy, Kesanupalli (Post), Narasaraopet (Mandal), Palnadu Dist.- 522601.
Hughes Syndrome is also known as Antiphospolipid syndrome (APS) is an autoimmune disorder characterized by hypercoagualability, leading to venous and /or arterial thrombosis and pregnancy-related complications. Obstetric manifestations include recurrent miscarriges, fetal death beyond the 10th week of gestation, and/or premature births. The clinical criteria for APS encompasses several specific events : three or more consecutive, unexplained miscarriages occurring before the 10th week of gestation; one or more unexplained deaths of a morphologically normal fetus at or beyond the 10th week; and one or more premature births of a morphologically normal neonate before the 34th week of gestation, typically due to eclampsia, severe preeclampsia, or placental insufficiency. From a serological perspective, the standard laboratory tests for APS involve the detection of a lupus anticoagulant(LAC) or IgG and/or IgManti-ß2 -glycoprotein antibodies (anti-ß2-GpI). For diagnosis, these antibodies must be present on two or more occasions at least 12 weeks apart.According to the international Consensus Criteria (Revised Sapporo Criteria) , a diagnosis of APS requires the presence of at least one clinical and one laboratory criterion. Treatment of APS primarily involves anticoagulant and antiplatelet therapy, typically requiring lifelong anticoagulation.In cases of pregnancy morbidity, low-dose aspirin combined with low molecular weight heparin (LMWH) Is considered the standard treatment. In rare and severe cases such as catastrophic antiphospholipid syndrome, aggressive management with anticoagulation, plasma exchange, and corticosteroids is warranted. The average age of patients with primary APS is reported to be around 35 to 40 years and the conditions is significantly more common in women than in men..
APLA were first described by conely in the 1950’s when it was noted that patients with lupus often had prolonged activated partial thromboplastin times(APTT).The original description of the syndrome was made by Graham Hughes in 1983[1], with SLE and LAC date back to late 1950s[2-3] single vessel involment or multiple vascular occulusions may give rise to a wide variety of presentations in the APS. Any combonitions of vascular occulusive events may occur in the same individual and the time interval between them also varies considerably from weeks to months or even years. Prevalence of the APL in the general population ranges between 1 and 5%. However ,only a minority of these individuals develops the APS. Some estimates indicate that the incidence of APS is around 5 new cases for 100,000 persons for year and the prevealence around 40-50cases for 100,000[4] in subset of patients, thrombosis can involove stimultaneously multiple organs, conifiguring the so-called “catastrophic antiphospholipd syndrome”(CAPS)
Epidemiology:
Antiphospholipid antibody are not specific ofAPS and can be found in healthy individuals.Nevertheless, the prevalence of APL postitivity and APS inn the general population has not been extensively analyzed and only two epidemiological population-based studies have been performed so far. In the first one, the authors studied and epidemiology of APS between 2000 and 2015an inception cohort of Olmsted country, minesota, through a recorded linkage system. The annual incidence of APS in adults aged>18 years was 2.1(95% confidence interval 1.4-2.80) per 100,000n population. Incidence rates were similar in both sexes. The estimated prevalence of APS was 50 (95% CI 42-58) per 100,000 population, and was similar in both sexes[5]. In the second study, performed in korea between 2007 and 2018, with date extracted from Health Insurance and Review Agency, an incidence of 0.75 per 100,000 person – year(95% confidence interval 0.73-0.78) was found, while the prevalence was 6.19 per 100,000 people.[6]
Thrombosis: The presence of antiphospholipid antibodies is risk factor for thrombosis The APS action group reported a literatrure review focused on the prevalence of antiphospholipid antibodies in the general population with preagnancy morbidity, strocks, myocardial infraction and deep vein thrombosis. The authors estimated that -13% of individual with stock,-11% of individual with myocardial infraction and -9.5% of individual with deep vein thrombosis are positive for antiphospholipid antibodies[7]. Another study in women <50years of age who had a strock showd that 17% were positive for lipus anticoagulant compared with 0.7% in the control group[8]. Positivity for lupus anticoagulant combaind with ostrogen-containing oral contraceptive use or smoking increased the risk further[ .
Pregnancy Complication: The APS ACTION group showed that 6% of patients with relavent pregnancy morbidity were positive for antiphospholipids antibodies[10]. Recurrent miscarriage is the most frequent complication and is observed in the majiorty(-54%) of women with obstetrical APS included in the European Registry Obstetrical Antiphospholipid syndrome11]. Fetal death is considerd to the consequence of placental dysfunction and is strongly associated with antiphospholipid antibodies[12-13]. In an analyasis of 512 stillbirths collaborative Research Network from 2006 to 2008, 11% (95%CI 8.4-14.4) of the women were positive for antiphospholipid antibodies[14]
Autoimmune Disease: Antiphospholipid antibodies can be detected in association with other systemic autoimmune diseases, most frequently SLE the prevaleance of antiphospholipid antibodies among patients with SLE ranges from 15% to 34% for lupus anticogulent, from 12% to 44% for anticrdiolipin and from 10% to 19% for anti-ß2- glycoprotein 1 antibodies. Of individual with SLE who are positive for antiphopholipid antibodies, 20-50% develops thrombotic events[15]
Etiology:
APL antibodies are present in a significant proportion of people with certain autoimmune or rheumatic disorders. The following are common autoimmune or rheumatic disorders and the proportion of patients having aPL antibodies (note that this is a percentage of patients, not the clinical state of APS
The following infections are linked to APS [16]
APS-related drugs include the following:
Pathophysiology:
It is recognized APL positivity is the most frequent acquired risk factor and is more realted to thrombotic events and gestational morbididty. Antiphospholipid antibodies(lipus anticoagulant, anticardiolipin and anti-ß2-glycoprotein-I) are classificatory antibodies of the diseases,used for diagnosis, but also important elements in the parthogenesis of APS. Althrough the presence of these antibodies is predisposing factor for thrombotic events, a second triggers, such as infection, prolonged rest or an inflammatory state, s usually necessary for the progression of the syndrome . Antiphospholipid antibodies bind to phospholipids and plasma or membrane proteins expressed in various cells (platelets, endothelial cells, monocytes, fibroblastes and trophoblastes),producing a prothrombotic state. Despite the know thrombophilic action of antiphospholipid antibodies, the exact pathogenises of the diseases is not yet fully elucidated.ß2-glycoprotein-I(ß2GPI) and prothrombin appear to be the major binding proteins in these antibodies involved in the pathogenesis of the disease. There is also a gentic component related to the HLA class2 system that needs to be better studied, and which may predispopose the individual to the disease.[17-18]
The pathophysiology of Antiphospholipid syndrom
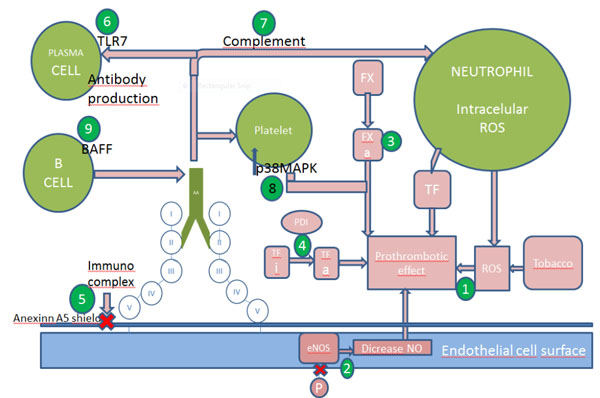
Mechanisms of thrombosis and possible targets. Numbers indicates the sites of action of the following drugs: 1N-acetilcystein, 2stains,3 hydroxycholoroquine ,fluvastain and FXa inhibitors(anticogaulents e.g. low molecular weight heparin ,fondaparinux, rivaroxaban and edoxaban),4 PDI inhibitors and hydroxycholorquine, 5hydroxychloroquine, 6 TLR7 inhibitors, 7 heparin, eculizumab, 8 belimumab
Diagnosis:
There is no single test can screen a patient for APL. One must perform the entire panel on patients suspected of having APLA. The panel would include
There are many confounding factors when teasting forAPLA. One is the there is ahigh ratye of false postivites with acute thrombosis-especially lupus inhibitors. Most patients who have only an isolsalted positive lupus inhibitor at the time of diagnosis will on repeat testing will have negativite testing. Second, many patients will have low titers tests, especially anticardiolipin assys so only titers >99th percentile or anticardioplipin antibody titers >40units are significant. Also the direct oral anticoagulants (DOACs) interfere with all coagulation based testing. Currently the Sydney criteria are used to diagnoses APLA syndrome-this requires both clinic and laboratory findings
Laboratory:
One or more positive tests repeatedly positive when tested at least 12 weeks apart:
*lupus inhibitor
*Anticardiolipin antibody- greater than either 99th percentile or greater than MPL or GPL units
*Antibeta2glycoprotien –greater than 99th percentile
Triple Positive-Patients are those with all three tests positive. These patients appear to be at high risk for thrombosis and at higher risk of “breaking through wafarin”. Occasional patients are seen who consistently have negative laboratory testing for APLA but have many of the clinical features of APLA such as thrombocytopenia, thrombosis and miscarriages. It is probable that these patients do have “APLA- negative APLA syndrome”and they should be treated as such.[19]
Clinical Manifestation: The main clinical manifestations of APS are the occurrence of thrombosis(arterial and /or venous) and / or preagnancy morbidity, including recurrent miscarriages, featl deths and late preagnancy, strockare frequent complications such as pre-eclamsia and intrauterine growth restrication . in addition, APS can be asspciated with a wide variety of other clinical symptoms
Management:
Although there are few prospective trails of therapy in APL, several lessons may be learned from retrosepective. While APLA does apper to be an autoimmune disease, immunosuppression does not prevent recurrent thrombosis, featl loss, or neurological syndromes. Therefore, immunospperssions should not play a role in the therpy of thrombotic APLA. The only exception to this is “catastrophic APLA” were plasmapheresis and immunosuppressions playas a crucial role. It used to be thought that anticoagulatuion with wafarin to an INRof 3.0-3.5 was effective in patients with APLA. However, randomized trails demonstrated that an INR range of 2.0-3.0 is just as effective as the higher INR range- at least for venous diseases. As mentioned above, some patients will fail wafrin and will require more aggressive anticoagulation. [20]. Controversial remains about treatment for arterial diseases-some recommend INR 3.0-3.5 while there is date showing INR 2-3 plus asprin may be effective. The role of DOAC is unclear. A clinical trail show DOAC were inferior to warfarin in “triple positive” patients and are contraindicated. For select patients with single positive APLA DOAC may be a consideration if there are challenges with warfarin.[21]
Thrombocytopenia:
Thrombocytopenia in patients with APLA occursin those patients prone to thrombosis due to activated platelets expressing the epitopes for APLA. The low platelet counts makes anticoagulation hazardus. In addition , patients with APLA are often high surgical risks. Thrombocytopenia may responded to steroids, immunoglobulin and IV-anti-D. Danazol 200mg po QID is effective in many patients with APLA realted thrombocytopenia as well as rituximab
Primary Prophylaxix:
There is no evidence to support primary prophyloxis of persistently positive APL patients should without thrombotic or gestational events. However, these patients should receive prophylactic heparin in situations of high thrombotic risk such as immobilization, hospitalization or postoperative. These patients should also avoid situations that increase the risk of thrombosis such as somking or the useof estrogen[22]. LA is the test with the highest predictive value for thrombotic events and unfavorable gestational outcomes, and its positivity should be considered when introducing prophylaxis.[23]
Secondary Prophylaxis:
After the acute episode,longe-term treatment with oral anticoagulant is therapy of choice. The recommened 2.0and 3.0 and for arterial phenomena, between 2.5 and 3.5. recents studies have found that higher INRs, between 3and 4, added an increased risk of bleeding[24].in addition to anticoagulant therapy , hydroxychloroquine seems to reduce APL titers and has beneficial antithrombotic role for patients with APS. Therefore, it should be added in all patients with APS associated with SLE.[25] In the specifc case of a first epiode of atherpsclerotic ischemic CVAin low-risk patients, treatment with asprin 300mg or double antiplatelet therapy may be attempted before anticoagulation is indicated. In case of thromboemoblic ischemiaCVA the treatment of choice is anticoagulants. It is not yet known whether non-vitamin k antagonist oral anticogalent (Xa-factor inhibitors and direct thrombin inhibitors) are effective in the treatment of APS. Clinical trails are being conducted to assess the actual benefit in thise subpopulation of patients.[26-27] . nevertheless, several cases reports of thrombiotic events in APS patients taking this new oral anticogalants were recently published[28]
Treatment of catastrophic APS: Patients with CAPS, in addition to anticoagulation with heparin, should recives immunosuppressions with corticoid heparin, should (prednisone,1mg/kg/day) or pulses of methylprednisolone associated with plasmapheresis or intravenous immunoglobulin. Rituximab can be used in refractory cases[29]. Recent studies have demonstrated benefits from the use of anti-C5 monclonal antibody (eculizumb) in patients with CAPS, providing the importance of complement activation in cases of thrombotic microangiopathy.[30]
CONCLUSION:
In conclusion, it is evident that a variety of mechanisms may act independently, or in conjuction, to lead to the clinical features associated with APS. The challenges is to determine with venous and/or arterial thrombosis, and pregnancy loss, as well as the key mechanisms in the non-criteria manifestitions ofAPS.
REFERENCES



Dr. Manchineni Prasada Rao*, Dr. V Rajini, Dr. Y Narasimha Rao, Kattimanda Jaya Prakash, A Review on Hughes Syndrome, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 581-588. https://doi.org/10.5281/zenodo.15804664
 10.5281/zenodo.15804664
10.5281/zenodo.15804664