
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Xavier Pharmacy College, Jajpur, Odisha
Recent progress in artificial intelligence (AI) changes drug discovery, allowing for the design and optimization of therapeutic agents with small molecules. Using deep learning and generative models of, researchers can explore larger chemical domains (estimated as ~1060 drug-like compounds) more efficiently than traditional methods. This essay features AI-controlled molecular design approaches including deep generation architectures (RNNs, VAEs, GANs, flow normalization, diffusion models, ground networks and transformer models). Finally, we highlight future directions such as the, including interpretable and fewer generation models, large-scale preparation with speech models of chemistry, and tighter integration into experimental workflows of AI. This comprehensive overview is based on the latest literature to assess the current status and promise of AI-regulated molecular design in discovering drugs.
The Drug discovery of active substances is traditionally slow, expensive and unstable, often requiring billions of dollars in the market. For example, the development and verification of a single drug candidate costs around USD 2.5 billion over a decade. An important bottleneck is examining the chemical space to identify new "drug-like" compounds with the correct balance of properties. Over the last years, the acceleration of this process has increased in an AI-controlled manner. was used to actually confirm biological activity, predict biological activity, design new scaffolds, and optimize the pharmacokinetic properties. The 2022 analysis found that the first candidates for AI-designed drug. Achieved clinical research in 2020, and by 2022 dozen companies (e.g., Insilico Medicine, Exscientia, Schrödinger) had AI candidates in Phase I development. The estimates show that over AI-controlled discovery programs are underway in human experiments with approximately 15 molecules, reflecting the rapid growth of this field. Therefore, AI is placed as a destructive force. This overview checks the main rate of AI and optimization of de-novo molecular design. This will highlight the literature from 2020 to 2025. These methods are explained in AI-designed case studies of connections and applications, explain the current challenges and limitations of, and outline future research directions.
BACKGROUND:
Traditional active substance discovery is based on experimental screening and established arithmetic tools such as the quantitative structure-activity ratio model (QSAR) and as molecular docking. Although QSAR and machine learning have been used for a long time to predict biological activity from known chemical types, these methods typically search within existing chemical spaces. For example, the early QSAR model and the high dock of the could triage large libraries with high throughput, but there was the risk of new scaffolding. The molecular docking program (e.g. Autodock Vina) evaluates the interaction between ligands and targets, but the throughput and accuracy of Traditional docking is limited. These approaches are often guided by drug-like heuristics (such as Lipinski and QED rules) to ensure candidate validity. As a result, traditional pipelines tend to be themed about known connections or analogs. AI changes the paradigm by enabling the De-Novo design to create an entirely new chemical structure from scratch. The Deep-Learning model can directly learn the representation of molecules from data (e.g. diagrams or smile strings) and propose new structures that meet the desired limitations. This shift builds into previous computational chemistry and extends to generation modelling. It is important that AI-based methods can handle several goals and complex facility landscapes. For example, the generative model can propose candidates that not only match the target binding site but also simultaneously maximize stability, solubility, and minimum toxicity of. Furthermore, structure-based designs with progression such as Alphafold2 (2021) protein structures are now more predictable with near-experimental accuracy, even if the crystal structure is not available. In summary, the background of molecular design was developed to count chemical libraries and evaluate them to use KI models to create and refine new molecules with repeat agents that expand the pool of accessible candidate drugs.
AI TECHNOLOGY AND MODELS IN MOLECULAR DESIGN:
Molecular Design includes a variety of technologies. Overall, these methods in:
Below we will explain the most important model classes and expressions.
Molecular Representation:
The input of the AI model includes the string format of the molecule (smile/selfie) or graph-based representation, as well as 3D coordinate speed. Robust codes such as Selfies ensure the validity of the molecules generated. The representation of the diagram (atoms as nodes, tie as edges) naturally catches molecular structures and is often used in graph neural networks (GNNs). The new transformer-based "chemical language" model also works with talk smiles or graphics fragments for the de-novo generation.
Predictive Model (Performance Prediction):
Deep Neural Networks (DNNs) containing GNNs are in a database of molecules of known activities (binding affinity, toxicity, solubility, managers, etc.) for predicting properties. These models serve as a quick alternative to expensive experiments or physical calculations. For example, GNNs can be trained to predict ligand-target interactions or certification points. In drug design, such predictive ML models often form a "scoring" function that leads the way in which is generated.
Variational Autoencoder (VAEs):
Creates a new structure by creating VAEs and decoding (sample) that encode molecules in continuous latent space. In chemistry, graph-based VAEs (e.g. GraphVAE) and smile-based VAEs were used to generate new scaffolds. The impairment of the latent vector allows this model to interpolate between known molecules, allowing for the study of the chemical region.
Generating Partner Network:
GANS checks the generator network against discriminators that distinguish between the generated molecule and the actual molecule. In chemistry-informatics, geese such as the Molgan Molecular graph are generated by optimizing the generator and deceiving the discriminator. Geese can quickly generate different candidates, but careful training is required to avoid chemically ineffective or unstable molecules. In fact, there are known limitations to the fact that geese can use structures that cannot be chemically inferred if the training data are not different enough.
Normalization Flow:
These invertible models learn the allocation of biojuots between the nucleus graph and the latent space. Examples are GraphNVP, GraphAF, Moflow and related models for Flow. Invertibility allows for accurate probability estimates and scans. In other words, it is attractive for generating graphics. The current was extended to 3D by manipulating the position of the Atom.
Diffusion Models:
Inspired by the formation of a diffusion process, these models convey large molecules in realistic structures. Latest work has adopted the diffusion of molecular graph and 3D geometry. This provides modern performance in the production of conformers and molecular optimizations. The diffusion model of 3D molecular design can learn complex structure distributions and create molecules with specific physical limitations. Application includes prediction of docking poses (such as Diffdock) and production of DE-Novo-3d-League.
Sequence Model (RNNS/Trans):
The repeating neural network (RNNS) and the Trans architecture treat smiles as "chemical language." By learning token sequences, these models can create a real smile. Trans-based models (e.g. Smiles-Bert, Molgpt) are grown in a corpus of large molecules and show strong performance on tasks such as property prediction and molecules. A recent work ("Taiga") combined a transformer and reinforcement learning to distort production into desirable drug-like properties. The large-scale model trained with chemical data (token moles and similar) represents a new limitation encoding both the 2D topology and the chemistry semantics of learning for generation.
Reinforcement Learning (RL):
RL frameworks deal with molecule era as a sequential selection process, with a praise feature reflecting goal objectives (e.g. excessive binding score, low toxicity). Methods like policy-gradient or Proximal Policy Optimization had been used to navigate the latent area of a pre-skilled generator for asset optimization. For example, a method referred to as Mercator or the aforementioned Taiga makes use of RL on latent vectors to growth drug-likeness scores (e.g. QED). RL is likewise regularly hired to fine-song collection models, encouraging them to advocate molecules with better sports even as retaining validity.
Graph Neural Networks (GNSs):
Beyond property prediction, GNSs also acts as a component in the generative model. Figure transformers and message networks can be issued directly on a molecular diagram, atom-by-bond, or per bond (such as graphene vent). Such a GNN generator can create chemically structured proposals and condition them for the desired attributes. Your Native complement sequence model to ensure consumption of molecular graphs.
Hybrid and Multimodal Models:
Many contemporary-day tactics integrate techniques. For instance, deep interactive studying integrates a graph transformer (encoding target–ligand interplay networks) with a chemical language version to enable “zero-shot” layout of ligands for brand spanking new targets. Other strategies fuse image-primarily based total representations, textual descriptions, or protein systems into the generative process. Multimodal deep studying – merging chemical graphs with protein pocket information – is a lively area. These AI strategies may be summarized as illustrated in Figure 1. Notably, deep generative architectures span VAEs, GANs, normalizing flows, diffusion networks, graph-primarily based totally mills, and sequence-primarily based totally transformers. Each has strengths: RNN/transformer mills excel at SMILES-primarily based totally layout; VAEs and flows offer controllable latent spaces; GANs produce excessive diversity; diffusion and flows are well-applicable for 3-D structure; and GNNs clearly cope with graph constraints. In practice, those fashions are coupled with scoring features or RL to manual molecules in the direction of more than one objective (e.g. target affinity, novelty, synthesis potential). For example, the dismantled potential Vaes allow for axes sampling to accommodate properties such as biological activity and solubility. In summary, the AI toolbox for molecular design is rich and allows for new strategies to grow and navigate the chemical domain.
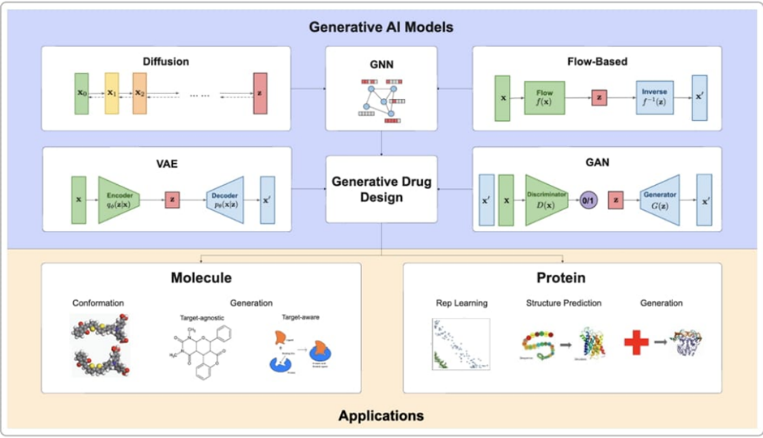
Figure 1. Generation AI model for molecular design. Latest approaches include (top column) diffusion model, graph neural networks (GNNs), normalized electrical models, variation car codes (VAEs), and generation controversial networks (GANs), chemical and latent structures, and (bottom row) applications new structures or (subformed) (Tang et al., 2024 [84†]).
CASE STUDY AND APPLICATION:
The potential of AI in the discovery of drugs is illustrated in several top-class examples:
Novel Antibiotic "Halicin" (2020):
Stokes et al. Neural networks were trained with curated data records of 2,500 molecules to predict broad spectrum antibacterial activity. They examined 100 million connections in Silico and identified halicin, a previously uncharacterized molecule. This demonstrated an efficacy of against many resistant bacteria in-vitro and in Mouse infection models. The discovery of halicin showed that AI was structurally discovered in a new way of traditional chemical intuition beyond.
PPARy & Ligands Via Deep Interactome Learning (2024):
Schneider and his employees have developed chemical language models with AI methods combined with graph ant trans and specific target profiles. This model was applied to the growth factor-activated receptor gamma (PPARy) of peroxisome, and the model generated several candidate leagues. The team synthesized several first-class designs, experimentally validated as powerful partial agonists with the intended selectivity of X-ray crystallography confirmed the predicted binding mode Successful suppression in Silico to lab translation.
AI-Specific Clinical Candidates:
The industry has announced several first-class candidates discovered through AI. For example, Ex Scientia DSP-1181 (serotonin receptor agonist of OCD) in Phase I. Similarly, insulin sensitivity and neurological candidates for Insilico Medicine achieved a test of Todd Wills (CAS) points out that around 15 AI-derived molecules were, clinical development until 2022. Such examples demonstrate that pharmaceutical companies view AI as a viable tool for lead detection.
Virtual Screening and Docking Enhancement:
AI is also used in virtual screening. In the example, on a diffusion base, we use the diffusion model learned using Diffdock (/Abdul Latif Jameel Clinic) to create candidate leagues in protein binding bags. Diffdock achieved a higher accuracy of to dock benchmarks as a traditional tool by implicitly modelling the ligand flexibility of ligand. Reports show that ligands (within 2 Å) are docked correctly in about 22% of cases, exceeding the old method. Such progress promises to accelerate the hit identification phase by quickly narrowing the connection that probably joins the Target.
Protein Structure Prediction (AlphaFold2, 2021):
The success of small molecules not directly constructed has the success of AlphaFold2 in dissolving protein structures with a deep impact on drug design. Alphafold2 (DeepMind) predicts 3D structures from amino acid sequences with a crystallographic accuracy of nearly. At the beginning of 2023, the database of AlphaFold protein structures ~200 million predicted structures (covering almost all known proteins). These models allow medical chemists to visualize the target and perform structure-based virtual screening. This can even be done with "and tunable" proteins. The impact on molecular design is important. Drug candidates can now be modelled at predicted binding sites.
Multimodal and Peptide Design:
Other uses include AI for peptide and protein therapeutics. For example, a trans-based model trained with a protein sequence can create new antibodies or peptide candidates. The Token-Mol model (Wang et al., 2025) illustrates an LLM approach for 3D demonstration molecule production. In fact, companies have used AI to design candidate biology such as: Although detailed case studies have been conducted, these areas quickly provide extensions of "molecular design" beyond small molecules. These cases show the promise of AI algorithms. The algorithm actually created new chemical types (for example, halicin) and accelerated the pipeline (searching millions of connections reduces searches to fewer leads). We also emphasize the importance of verification. Experimental tests were conducted on all molecules designed by Synthetic and AI to confirm activity. Surprisingly, in the case of PPARy we predict experimental efficacy. Such success suggests that the combination of AI, chemical knowledge and high-throughput experiments can reduce cycle times. However, it is clear that AI results often require people and labs to be refined before candidates can proceed.
CHALLENGES AND LIMITATIONS:
Despite advances, several challenges dominate the hype around AI in molecular design. Main Edition:
Data Quality and Coverage:
Most AI models rely on existing data (known molecules and bioactivities) in training. If these data records are skewed to a particular chemistry type or target, the model can only generate similar connections. Like Crucitti et al. It should be noted that current AI is approaching "optimizing molecules in an established chemistry room" and has acquired the ability to invent truly new structures. In fact, data records are often recorded under complex scaffolds or rare drug belts. This "training settings" limits the novelty. The model does not replicate the well-known motifs or suggest alternatives.
Limited Chemical Variability:
Many hits designed by AI to expand the above are similar to drug or lead connections. For example, analysis of early AI candidates (excientias DSP-1181; discovered that they share a core scaffold with existing molecules. GANs and other generators can even produce chemically unstable or syntactically ineffective molecules when the training set does not have a variety. Although necessary to overcome these required data records and new generation goals, is still an open issue.
Synthetic Feasibility:
AI can propose molecules to synthesize with silico, but it can be difficult or expensive to. Allowing the generated candidates to be chemically synthesized is a major hurdle. Generative models often require custom synthesis of new backbones. This is more intensive for resource-intensive 1-than known order connections. Although some progress has been made (for example, which includes rules based on synthetic accessibility or reactions in production), in fact, many AI results still require medical chemistry effort. If you cannot create or scale connections, you can disable excessive optimal activity prediction.
Computational Cost:
Some AI methods (especially 3D models and large transformers) require a considerable amount of computing power. End-to-end pipelines (generated + virtual screening) can exceed the cost of a simpler approach unless carefully stopped. For example, structure-based filtering of the AI-generated library for molecular docking or ML-based affinity prediction is dependent on the computing step. The balance of innovation over arithmetic resources is a continuing problem of. In which scenario, can AI really save time and money compared to standard screening for large virtual libraries? The answer is probably dependent on the context of the project and is topics of active discussion.
Interpretability and Trust:
Deep learning models are often "black boxes." Medical chemist needs to understand why the model proposes a specific molecule. However, is still difficult. In general, users cannot simply track the functions that led to design within their data. As a result, AI proposals can even be confronted with experimentally validated scepticism. Junaid (2025) shows that assurance of data consistency and interpretability is a "big challenge" for AI in biology. In practice, AI control projects generally require wet mark checking to generate trust. The need for experimental verification means that AI is more of an assistant to human experts than replacements.
Reproducibility and Validation:
While many AI studies have reported impressive reports on Silico results, leads to successful medication. Sceptics have shown that arithmetic metrics (e.g., docking scores) do not always correlate with experimental results. requires a standardized benchmark and practical attempts using AI suggestions. Several efforts, such as Moses, have provided benchmarks for the generative model, but the city standards are still arising. Until, other AI-specific molecules exhibit clinical benefits, with questions about reproducibility and efficacy remaining.
Scope of Application:
AI methods are often characterized by well-interrogated destinations. Designing molecules for or complex targets (such as specific enzymes or powerful proteins) remains a challenge. Additionally, designs with some targets or polyphor malacologist (after connections that modulate several paths) add complexity that simply does not handle many current models. Crucitti et al. Observe the fact that it is unclear whether Ki can actually implement the DE-Novo design of the target, or whether it is always based on an analogue of well-known ligands. The Ju umpire is not yet able to create candidates for the AI "dark goals outside the appropriately mentioned chemical domain.” In summary, AI-controlled molecular design unlocks new possibilities, but also highlights the essential limitation. The model depends on the data and your costs are as good as the entry. The challenges and verification requirements for Synthesis mean that AI will rather replace traditional chemistry in a short period of time. A careful hybrid approach combining AI exploration, expert management, and experimental feedback is essential.
FUTURE DIRECTION:
There will be some trends to address these challenges over the next few years.
Interpretable and Controlled Models:
The focus on the structure of the generated model, which provides a specific level of explanation. Techniques such as relaxed VAEs (where the latent dimensions correspond to a particular characteristic) and attention-based GNNs can help to illuminate the decisions of the model. For example, the method arising from the announcement of the latent factor "controllable production," can affect predictive activity. Future research focuses on generation frameworks for white boxes or post-declaration tools or up to generation frameworks to make AI proposals more transparent.
Few-shots and Transfer Learning:
Many problems with drug design have limited data (rare diseases, new pathogens). Techniques learned from a few examples are extremely important. Data expansion (e.g., smile randomization) and transfer learning (a fine chemistry language model for small data records) have already been used to extend the model to a low data regime. In the Zero-Shot learning approach, the model uses side information (target properties, text description) to generate an invisible class of molecules. As a larger, preceded by the molecular model (similar to GPT for chemistry), adjustment to tasks below is a critical direction.
Large Language Models and Multimodal Learning:
Large-scale pre-educational models trained on large chemical and biological datasets (millions of connections, protein sequences, literature) are feasible. Latest works such as Token-Mol show LLMS that integrate 3D structural information into the production process. In the future, more mergers of the modalities are expected. For example, a combination of chemical smiles and protein structures or bioactivity descriptions in text is an explanation of biological activity in a single model. Such a basic model could be better generalized for, allowing for the production of molecules informed by biology (production of inhibitors based on protein sequence). The success of LLMS in NLP suggests that a similar revolution in chemistry is possible as soon as the model is large enough and trained for different data.
Active learning and closed-loop design:
A promising orientation is the tight integration of AI in experimental laboratories. An automated synthesis and screening platform ("Autonomous Driving Laboratory") can quickly test connections generated by the-AI and supply results again with the model. Active learning strategy, speeds up the discovery cycle as the model is repeatedly numerators in the query (maximum information gain). For example, AI can synthesize small amounts of different candidates. After testing, the results improve the predictive model and suggest a better molecule. This approach with a closed loop uses experiments efficiently and is already piloted by several consortiums.
Better objective function:
Improved property evaluation and integration is a significant. Instead of optimizing a single point (such as a dock), future models better compensate for several goals, such as potency ADMET, novelty, and ease of synthesis. Advances in multi-lens optimization (Pareto Front Method) allows chemists to find compromises between competitive factors. Furthermore, including actual limitations (patent landscape, market needs) via AI control cost functions is an ambitious field.
Ethics, Bias, and Cooperation:
When AI tools grow, it is important to monitor distortions and ensure data protection (for example, your own combined library). The AI regulatory framework and the best practices in F&E could be developed. Academic and industry-related industry collaboration on parts of the benchmark, data records (in terms of IP), and parts of the open-source tool. Overall, this field matures by combining innovation and community standards. Overall, the future of AI in molecular design is to build more intelligent and generalizable models that work hand in hand with chemists. Advances in interpretability, large-scale pre- and experimental integration will help you recognize the promise of faster, cheaper drug discovery.
CONCLUSION:
AI has begun a new era of molecular design in active substance discovery. Over the past five years, deep learning and generative models have expanded the toolkits available to medical chemists, allowing rapid research into the chemical domain, and enabling innovative drug candidate proposals. Success stories such as halicin and AI-generated PPARy Agonists show that mechanical intelligence can complement human creativity. At the same time, this field appears important: data limits, feasibility of synthesis, and the need for verification that AI is not a magical ball. Current Method often finds variations on well-known topics and is not a completely unprecedented solution. Therefore, AI in molecular design may remain an augmentation technique - however, accelerated the hypothesis and prioritization of leads is still based on expert monitoring and experimental testing. Explainable ki development, a small number of learning and multimodal foundation models promise that will further integrate AI. Ultimately, the most successful application comes from close collaboration between computer scientists and laboratory researchers. Using AI strength in pattern recognition at and searching along with human insights and experimental feedback on the drug discovery community hopes to tackle the difficulty of providing new treatments so far more efficiently.
REFERENCES






: Sai Swagatika Das, Tushar Kanti Das, Biswa Bhusan Padhi, Nityapriya Maharana, Jeeban Pradeep Agnihotry, Chandrakanta Das, AI in Molecular Design and Optimization in Drug Discovery, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 373-382. https://doi.org/10.5281/zenodo.15791238
 10.5281/zenodo.15791238
10.5281/zenodo.15791238