
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,2 Priyadarshini J. L. College of Pharmacy, Hingna, Nagpur-440016, Maharashtra, India.
3,4 K. C. Bajaj College of Pharmacy & Research, Jaripatka, Nagpur-14, Maharashtra, India
5 Dr. Rajendra Gode College of Pharmacy, Malkapur-443101, Maharashtra, India.
6 Central India College of Pharmacy, Nagpur, Maharashtra, India.
Alzheimer’s disease (AD) is the most common neurodegenerative disorder and the leading cause of memory loss worldwide. Its prevalence is increasing at an alarming rate, largely due to the aging global population and improved life expectancy. As a result, AD has become a major public health concern, placing a substantial burden on patients, caregivers, and healthcare systems globally. Alzheimer’s disease accounts for approximately 60–70% of all cases of progressive cognitive impairment and dementia in older adults. At the biological and pathological level, the condition is marked by the buildup of ?-amyloid plaques outside nerve cells and tau-based neurofibrillary tangles within them. These harmful protein deposits interfere with communication between neurons, damage synaptic activity, and lead to ongoing loss of brain cells. We still don't fully understand the precise mechanisms that underlie Alzheimer's disease genesis and progression. Numerous theories have been put forth to explain the pathophysiology of the amyloid cascade hypothesis, tau propagation theory, neuroinflammation, oxidative stress, mitochondrial failure, and vascular involvement are all examples of neurodegeneration in AD. Increasing evidence suggests that Alzheimer’s disease results from a complex interaction of genetic, environmental, and lifestyle factors rather than a single causative mechanism. Early and accurate diagnosis of Alzheimer’s disease remains a significant challenge. Current diagnostic approaches rely on clinical assessment, neuropsychological testing, neuroimaging techniques, and cerebrospinal fluid biomarkers. Advances in biomarker research and molecular imaging have improved diagnostic accuracy; however, widespread implementation remains limited. This review seeks to summarize the present state of Alzheimer’s disease diagnosis and to explore emerging approaches as well as future directions for addressing the challenges posed by this condition.
The reason behind causing of dementia is Alzheimer's disease (AD), a progressive neurological disorder. Memory loss is one of the hallmarks of dementia, along with cognitive, linguistic, and problem-solving deficits. Globally, Alzheimer’s disease ranks as the sixth leading cause of death. Its growing prevalence places a substantial burden on healthcare systems and social support services, both directly and indirectly. A 2013 World Health Organisation report on the epidemiology of Alzheimer's disease states that the estimated 35.6 million people worldwide who had dementia in 2010 are predicted to quadruple by 2050. Dementia affects between 5–8% of people over 65 and rises to 25–50% for those over 85. The risk of dementia increases dramatically with age. Additionally, the prevalence of Alzheimer’s disease is reported to be lower in men than in women by approximately 19–29%. [1].
A definitive diagnosis of Alzheimer's disease needs a post-mortem analysis of brain tissue and positron emission tomography (PET) scans and the availability of biomarkers from cerebrospinal fluid (CSF) tests and, along with more recent clinical diagnostic criteria, can help identify the disease in living patients. Cholinesterase inhibitors, which can be taken at any stage of Alzheimer's dementia, and memantine, which is given for moderate to severe instances, are currently accessible treatments. These drugs have been demonstrated to improve quality of life for patients and their carers when given at the proper stages of the illness. Nevertheless, they do not alter disease progression or slow the overall rate of cognitive decline [2].
According to this hypothesis, in different parts of the brain, β-amyloid (Aβ) plaques develop and build up. These deposits, which are known as aberrant material which activates microglial cells and release inflammatory cytokines and causes neurodegeneration and neuronal death. Aβ plaques are made of Aβ peptides generated from amyloid precursor protein (APP). APP is breakdown by β-secretase, producing membrane-bound C-terminal fragments of 89 or 99 amino acids. The β-secretase that breaks APP at the β-sites Asp1 and Glu11 is called BACE1 (β-site APP-cleaving enzyme 1), known as Asp2 or memapsin-2. The Aβ?–?? and Aβ?–?? peptide isoforms are produced when γ-secretase breaks down the 99-amino acid C-terminal fragment. Under normal conditions, Aβ?–?? is the predominant soluble form of β-amyloid. However, alterations in the cleavage process can lead to the production of Aβ?–?? exhibits an increased propensity to aggregate and form plaques owing to the inclusion of two extra amino acids, isoleucine and alanine [3]. The mutations in the amyloid precursor protein (APP), presenilin 1, presenilin 2, or apolipoprotein E (APOE4) genes are primarily connected to changes in cleavage patterns. In addition to genetic factors, several neuropeptides are also believed to contribute to plaque formation. The combined accumulation of Aβ and tau proteins disrupts synaptic plasticity and ultimately results in neuronal cell loss [4]. Notwithstanding its broad acceptance, this idea remains contentious, as current research indicates that medications aimed at inhibiting amyloid plaque formation do not successfully halt or reverse cognitive deterioration. Consequently, there is a growing need to explore therapeutic strategies that target non-amyloid mechanisms, neuroinflammation, oxidative stress, tau pathology, and other contributing factors [5].
The microtubule-binding domains found in neuronal microtubule proteins called as tau proteins are needed for microtubule polymerization and stabilization, which shows cytoskeletal structure. The kinases that phosphorylate serine and threonine residues to initiate their interaction with microtubules contains cyclin-dependent kinase-5 (CDK5), Fyn kinase, and glycogen synthase kinase-3β (GSK3β). Out of these, CDK5 is important for the formation of neurofibrillary tangles. Excessive activation of CDK5 occurs when its regulatory subunit p35 is cleaved into p25, a process triggered by elevated cytosolic calcium levels. This abnormal activation results in excessive phosphorylation of tau proteins [6]. Excessive phosphorylation further reduces the ability of tau proteins to bind to microtubules. As a result, hyperphosphorylated tau aggregates into neurofibrillary tangles (NFTs) that accumulate within the cytoplasm and lose their capacity to support cellular structural integrity [7].
The Aβ is known to have antimicrobial properties. The neurones infected with conditions like HSV, Chlamydia, or spirochetes demonstrate increased in Aβ deposition and neurofibrillary tangles (NFTs). This shows that chronic, untreated infections may lead to Alzheimer's disease [8]. These infections initiate the immune system, and components of the innate immune system primarily Toll-like proteins play a crucial part in this process [9].
In most of the cases ‘Apo-Lipoprotein-E (APOE)’ gene is responsible for the incidence of AD. Other genes such as presenilin 1 and presenilin 2 are also associated [10].
The oxidative stress and mitochondrial dysfunction play an important role in the early cause of Alzheimer's disease. The mitochondrial impairment is responsible to decreased cytochrome C oxidase levels, and the permeability of the mitochondrial membrane may also be affected by oxidative stress-induced hyperactivation of glycogen synthase kinase-3 (GSK-3). These changes lead to an increase of reactive oxygen species (ROS) [11]. These ROS can be generated when metal ions, such as copper and zinc, adhere to Aβ plaques. The resulting oxidative damage affects the Aβ peptide itself, hindering its clearance, and also promotes lipid and protein oxidation in cell membranes, increasing their permeability and making neurons more vulnerable to degeneration [11, 12, 13]. Aβ plaques disrupt calcium storage in the endoplasmic reticulum, causing elevated cytosolic calcium levels. This increase contributes to glutathione depletion and excessive accumulation of reactive oxygen species (ROS) within cells. Via increasing membrane permeability and encouraging the production of both reactive nitrogen species (RNS) and reactive oxygen species (ROS), overactivation of N-Methyl-D-Aspartate-type glutamate receptors (NMDARs) may further elevate calcium influx. Moreover, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase can be directly triggered by Aβ proteins, resulting in the generation of free radicals [14].
Autophagy is the cell’s internal “housekeeping” or waste management system, responsible for clearing damaged proteins and dysfunctional organelles. The process begins with the nucleation of a structure called the phagophore, which engulfs the targeted cellular components. These autophagosomes then fuse with lysosomes to form autolysosomes, where the enclosed material is degraded and recycled. In Alzheimer’s disease, factors such as presenilin-1 abnormalities, oxidative stress, and tau neurofibrillary tangles can disrupt autophagic function, contributing to disease progression. [15, 16].
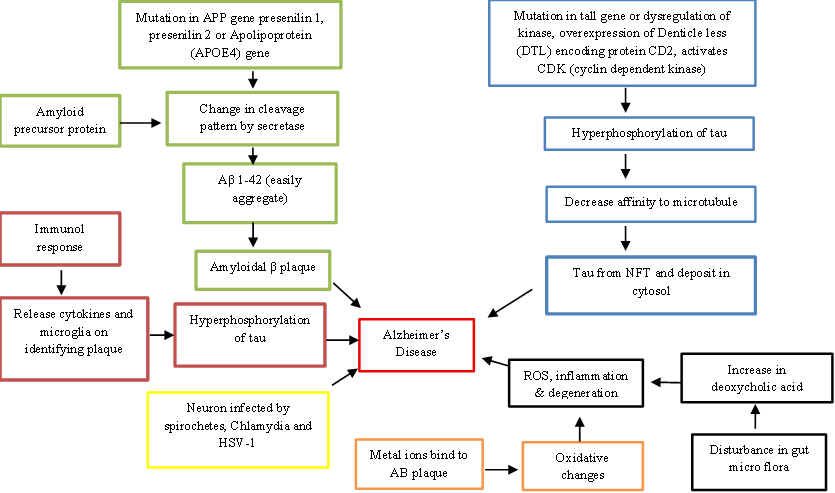
Figure 1: Mechanisms of Alzheimer’s Disease
To date, numerous attempts have been made to address and correct disruptions in neurotransmission. One key reason a specific treatment for Alzheimer’s disease remains unavailable is that the pathological changes associated with the disease often begin many years typically a decade before clinical symptoms become apparent.
2.1. FDA Approved Medicines
Alzheimer's disease medications now on the market mostly target cholinergic and glutamatergic neurotransmission, which simply relieves symptoms rather than slowing the disease's progression. Numerous clinical trials are underway to develop molecules that can target the underlying pathology, including amyloid and tau proteins [17]. Among approved treatments, three acetylcholinesterase inhibitors Donepezil, Rivastigmine, and Galantamine are used to enhance cognition by supporting cholinergic signaling. An FDA-approved medication called memantine is used to treat moderate to severe AD because it lessens excitotoxicity brought on by excessive glutamatergic activity. Recently developed medications for Alzheimer's disease are included in Table 1, although a number of them have not made it to market because of unsuccessful clinical studies.
Table 1: Medications created to treat Alzheimer's [18].
|
Drug Name |
Company |
Mechanism of Action |
Clinical Trial |
Reason behind failure |
|
Verubecestat |
Merck |
Inhibitors of β-secretase-1 (BACE-1) |
EPOCH Trial APECS Trial |
Showed no significant disease-modifying effect in AD |
|
Lanabecestat |
Astra Zeneca and Eli Lilly |
BACE 1 inhibitors |
AMARANTH and DAYBREAKALZ |
Failed to pass the interim futility assessment |
|
Atabecestat |
Janssen |
BACE 1 inhibitors |
EARLY |
Liver toxicity |
|
Solanezumab |
Eli Lily |
Amyloid-β–specific monoclonal antibody |
EXPEDITION III (PHASE III) |
Failed to show improvement in cognitive outcomes |
|
Azeliragon |
vTv Therapeutics |
Inhibitors of the receptor for advanced glycation end products (RAGE) |
STEADFAST (PHASE III) |
Lack of efficacy |
|
Pioglitazone |
Takeda and Zinfandel Pharmaceuticals |
Peroxisome Proliferator-activated Receptor |
- |
- |
|
Idalopirdine |
Lundbeck and Otsuka |
5HT6 antagonist |
STARSHINE, STARBEAM, STARBRIGHT (PHASE III) |
Showed no significant cognitive benefit |
Owing to the high failure rate of Alzheimer’s disease drugs, researchers are focusing on developing new agents aimed at providing symptomatic relief. A list of such drugs currently undergoing clinical trials is presented in Table 2.
Table 2: Some important drugs which are currently in clinical trials
|
Drug Name |
Company |
Mechanism of Action |
Clinical Trial |
Symptoms to be eased |
|
Brexpiprazole |
Otsuka and Lundbeck |
D2 partial agonist |
PHASE III |
Agitation |
|
Tetrahydrocannabinol |
Johns Hopkins University |
Cβ1/Cβ2 partial agonist |
PHASE III trials |
|
|
Nabilone |
Sunnybrook Health Sciences Centre |
Semisynthetic cannabinoid derivative |
Small scale PHASE III |
|
|
Pimavanserine |
Acadia |
Selective 5-HT2A serotonin inverse agonist |
PHASE II/III |
Psychotic |
|
Suvorexant |
Merck |
Dual antagonists of the orexin receptors |
PHASE III |
Insomnia |
|
Lemborexant |
Eisai |
Dual antagonists of the orexin receptors |
PHASE II |
Immunotherapy approaches are being explored to develop agents targeting amyloid and tau proteins. Additionally, alternative strategies aimed at addressing misfolded tau and promoting neuronal regeneration are currently under investigation. [18].
Gene therapy approaches for Alzheimer’s disease focus on identifying defective genes, DNA, or proteins and introducing corrective genetic material to restore normal cellular function. This is typically achieved by delivering new genes into living cells using viral vectors. For example, in a study involving ten patients with early-stage AD, the nerve growth factor (NGF) gene was inserted either ex vivo or in vivo to test its potential to support degenerating neurons [19]. In a different animal investigation, the PGC1-alpha (Peroxisome proliferator-activated receptor gamma coactivator 1-alpha) gene was inserted into the brains of mice using a modified virus. This intervention reduced Alzheimer’s-like pathology, improved memory, preserved neuronal populations, and decreased amyloid plaque formation compared to untreated mice [20]. Adeno-associated viral vector serotype 2 delivering NGF (AAV2 NGF) was recently tested in AD patients in a randomised clinical trial. Despite the lack of clinical advantages, the study verified that the treatment was both practical and well-tolerated [21].
Several hypotheses have been proposed to explain the mechanisms of Aβ immunotherapy. The primary actions involve antibody-mediated neutralization and solubilization of Aβ plaques, both in the brain and peripherally. Antibodies can also attach to amyloid seeds in the early phases of amyloid production, stopping subsequent aggregation [22]. Additional processes include the "peripheral sink" effect, which helps remove A from the brain into the plasma, and direct rupture of preexisting plaques [23].
In recent times, monoclonal antibodies are under research in clinical studies, including bapineuzumab, solanezumab, gantenerumab, crenezumab, and ponezumab. ARIA-E (vasogenic oedema) or ARIA-H (hemosiderosis and microhaemorrhages) are the examples of amyloid-related imaging abnormalities (ARIA), are among the side effects of several of these treatments [24]. A summary of therapeutics based on active and passive immunization strategies is demonstrated in Table 3.
Table 3: Active and Passive immunotherapeutic [25]
|
Drug Name |
Mechanism of Action |
Clinical Trial Phase |
|
Active Immunotherapeutic |
||
|
ACI-24 |
Induce anti-amyloid-β antibody production without activating inflammatory cellular responses. |
PHASE I |
|
ACI-35 |
A liposomal vaccine engineered to generate anti-phosphorylated tau antibodies. |
PHASE I |
|
AADvac-1 |
Contains the peptide KDNIKHVPGGGS designed to elicit anti-tau antibodies. |
PHASE III |
|
CAD106 |
A viral vector vaccine engineered to generate anti-Aβ immunity while minimizing T-cell activation. |
PHASE II |
|
Passive Immunotherapeutic |
||
|
Aducanumab |
Monoclonal antibody against Aβ. |
PHASE III |
|
Gantenerumab |
Binds to Aβ and promotes phagocytosis through microglial activation. |
PHASE III |
|
BAN2401 |
Selectively binds to soluble Aβ protofibrils. |
PHASE II |
|
Solanczumab |
Interacts with monomeric and soluble, non-fibrillar forms of Aβ. |
PHASE III |
Synthetic substances that imitate natural proteins and interact with biological targets to create comparable or improved effects are known as peptidomimetics [25]. These compounds are intended to target human β-secretase as well as phosphorylated tau proteins [26].
Probiotics support the growth of beneficial gut microbes, with prebiotics providing the necessary nutrients and environment for their development. In animal studies, patients were given FRAMELIN, which is a combination of probiotics such Lactobacillus acidophilus lysates, Bifidobacterium longum, omega-3 fatty acids, and vitamins. One of the exercise therapies was given as treadmill-based running for set periods of time. The Morris Water Maze Test, Open Field Test, microbiome analyses, and Spontaneous Alternation Tests were used to evaluate the results. The study found that taking probiotic supplements along with regular exercise can help lessen the symptoms of Alzheimer's disease and halt its progression [27].
Future studies on Alzheimer's disease treatments will concentrate on treating the underlying pathological characteristics, which include amyloid-β (Aβ) plaques and neurofibrillary tangles made of tau proteins. The optimal moment to act and the best target to reduce neurological decline are still up for debate [28, 29]. Another strategy being studied is strengthening inter-neuronal connections to improve cognitive performance [30]. The EU/US Clinical Trials in Alzheimer's Disease Task Force conducted several clinical trials in 2016 to assess best practices for patient recruiting, retention, trial infrastructure, and outcome evaluations, including biomarkers and objective cognitive testing [31].
3.1. Anti-amyloid Therapy
The development of harmful amyloid plaques is one of the first stages of Alzheimer's disease, according to the amyloid cascade theory [32]. According to a 2013 study, these aberrant plaques cause tau proteins to become phosphorylated. These proteins are subsequently carried by microtubules to nearby neurones, where they eventually cause neuronal death [33]. Monoclonal antibody-based passive immunotherapy is one treatment strategy. In order to help remove abnormal Aβ from the brain, this method entails administering antibodies that precisely target it. By 2014, two such monoclonal antibodies were designed to remove plaques from Alzheimer's patients [34, 35].
By inhibiting the enzymes that produce Aβ peptides from amyloid precursor protein (APP) is another way to reduce Aβ plaques in the brain. Specifically [36]. Early human trials have indicated that the new chemical VERUBECESTAT has a favourable safety profile and can lower Aβ levels in rodent brains by more than 40 times [37]. Researchers found in 2014 that Aβ buildup in amyloid-producing mice was significantly reduced when a monoclonal antibody and a BACE1 inhibitor were combined [38]. Many experts think that targeting Aβ through both pathways could eventually lead to an effective treatment for Alzheimer's disease, even though there are presently no human trials investigating this combined medication [39].
The treatments which are used to lower tau accumulation are being developed since tau proteins are thought to be directly contribute to the symptoms of Alzheimer's disease [40]. Various tau-targeting vaccines have demonstrated promising safety and efficacy in animal studies [41]. In a recent small-scale human study, one anti-tau agent showed both a favorable safety profile and a positive immune response [42]. Table 4 summarizes the therapies and targets presently being studied [43].
Table 4: Novel therapeutics for Alzheimer’s disease
|
Target |
Drug |
Phase of Study |
Date of completion/completed |
Results |
|
β-Amyloid |
CAD106 |
PHASE II |
May 2024 |
- |
|
CNP520 |
PHASE II |
May 2024 |
- |
|
|
LY3002813* |
- |
December 2020 |
Effective |
|
|
Solanezumab |
- |
Terminated May 2017 |
Not Effective |
|
|
1812 |
PHASE III |
October 2024 |
- |
|
|
Ponezumab |
- |
June 2011 |
Not effective |
|
|
KHK6640 |
- |
December 2017 |
None yet |
|
|
BACE1 |
JNJ-54861911 |
PHASE III |
October 2025 |
- |
|
LY3202626* |
PHASE III |
December 2024 |
- |
|
|
LY450139 |
- |
April 2011 |
Not effective |
|
|
P-tau |
JNJ-63733657 |
PHASE II |
February 2026 |
- |
|
BIIB-092 |
PHASE III |
September 2024 |
- |
|
|
APP |
Posiphen |
PHASE I |
- |
- |
|
RAGE |
Azeliragon |
- |
Terminated January 2019 |
Not effective |
|
Retinoid receptor |
Acitretin |
PHASE III |
February 2024 |
- |
|
Bexarotene |
PHASE III |
February 2025 |
- |
The progression of Alzheimer’s disease symptoms is associated with extensive dysfunction in neural networks. The gamma oscillations, or high-frequency brain wave rhythms, are important for interneural communication in nearly every brain circuit [44]. The researchers at the Massachusetts Institute of Technology found that inducing gamma-frequency oscillations increased cognitive performance and decreased Aβ accumulation in an animal model of AD [45]. This effect was achieved by non-invasively entraining the mouse cortex to a 40 Hz rhythm using a photic stimulator.
Stem cell therapy is a novel technique which has shown great potential in treating patients with AD. “Astrostem” is a stem cell-based treatment, derived from autologous adipose tissue. The human mesenchymal stem cell treatment is evaluated in a phase I study.
However, direct implantation of neural stem cells is not attainable because of their limited sources and compulsive immunogenicity [46].
Lanabecestat (also known as AZD3293 or LY3314814) is an orally active BACE1 inhibitor that showed promising results in preclinical studies. The clinical trials for other BACE1 inhibitors, such as verubecestat (MK8931) and elenbecestat (E2609), were terminated in June 2018 due to a lack of efficacy [47]. The Lilly and AstraZeneca received FDA Fast Track designation for the medication in 2016, and it advanced to Phase 3 clinical trials that same year [48,49]. Umibecestat (CNP520), another BACE1 inhibitor intended for early-stage AD, also had its clinical trial terminated in 2019 due to cognitive decline observed in study subjects [50,51].
Aβ peptide synthesis is decreased when γ-secretase is inhibited. In healthy individuals, Lilly's non-selective γ-secretase inhibitor Semagacestat (LY450139) showed a dose-dependent decrease in Aβ generation [52]. Avagacestat, another γ-secretase inhibitor, did not substantially improve Alzheimer's patients [53,54].
The activation of protein kinases which are responsible for stimulation of α-secretase, it has been shown to reduce Aβ levels [55]. Etazolate (EHT 0202), a pyrazolopyridine derivative with anxiolytic properties, is considered a promising α-secretase activator; it has been reported to inhibit Aβ-induced cell death and alleviate symptoms of Alzheimer’s disease [56]. Green tea is the main source of epigallocatechin gallate (EGCG, Sunphenone), a polyphenolic flavonoid with anti-inflammatory and neuroprotective properties. According to experimental research, EGCG improves cognitive impairments in APP/PS1 animal models via increasing α-secretase-mediated cleavage of APP [57, 58].
Dementia is one of the syndromes that is characterized by cognitive and functional decline. The pathophysiology of AD cannot be entirely explained by the numerous ideas that have been put forth. In most cases, no medication can stop or reverse the disease's course because the cause is still unclear, even if some may temporarily reduce symptoms. Understanding the genetic origin and molecular foundation of Alzheimer's disease has advanced significantly in recent years. While drugs like memantine and cholinesterase inhibitors can improve memory and alertness, they have no effect on life expectancy or the general course of Alzheimer's disease. Dietary and exercise changes are the only therapies that have been demonstrated to lower the risk of AD and perhaps halt cognitive decline as first-line measures for all individuals, regardless of their cognitive status. Most of the immunotherapeutic are in phase 3 of clinical trials. It is still unknown how many of the more than 140 medication candidates, the majority of which are disease-modifying drugs, will successfully move on to later stages of clinical trials. Other methods, such as metal ion chelators, peptidomimetics, and the use of speech as a diagnostic indicator, are still being researched and have not yet been extensively used in clinical practice. However, more and more research is still needed further to explore the mechanisms of actions of medications in seeking a promising therapy for Alzheimer’s disease.
ABBREVIATIONS
|
AD |
Alzheimer’s Disease |
|
APOE4 |
Apolipoprotein E4 |
|
APP |
Amyloid Precursor Protein |
|
Aβ |
Beta amyloid |
|
BACE |
β-site APP cleaving enzyme |
|
FDA |
Food and Drug Administration |
|
LOAD |
Late Onset AD |
|
NFT |
Neurofibrillary tangles |
|
NMDA |
N-methyl D-aspartate |
|
RAGE |
Receptor for advanced glycation end products |
REFERENCES






Mayur Lohi, Shilpa Deshpande, Pooja Dhenge, Akshay Bidwaik, Shweta Rane, Meenal Watkar, Alzheimer’s Disease: An Overview of Pathophysiology, Diagnosis, and Emerging Strategies, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 2973-2986. https://doi.org/10.5281/zenodo.18377430
 10.5281/zenodo.18377430
10.5281/zenodo.18377430