
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
SIRT-Pharmacy SAGE University bhopal
The combination of Artificial Intelligence (AI) and pharmaceutical science is creating exciting changes in the way new medicines are discovered and developed. Significant developments in artificial intelligence and machine learning offer a game-changing prospect for pharmaceutical dosage form testing, formulation, and medication discovery. AI can help lower development costs. In addition to predicting the pharmacokinetics and toxicity of potential drugs, machine learning techniques aid in the design of experiment. By prioritizing and optimizing lead compounds, this capability lessens the need for expensive and time-consuming animal testing. Artificial intelligence (AI) algorithms that evaluate actual patient data and their personalized medical strategies, improving patient adherence and treatment results. The many uses of AI in drug discovery, drug delivery dosage form designs, process optimization, testing, and pharmacokinetics/pharmacodynamics (PK/PD) research are examined in this thorough overview. This review focuses on how AI helps speed up the process, improve accuracy, and reduce costs by addressing the problems faced in traditional drug development methods. We explore the different ways AI contributes especially in identifying disease targets, selecting the most promising drug candidates, and making clinical trials more efficient. AI tools like machine learning, data analysis, and predictive models are giving researchers powerful ways to find patterns in large datasets, which helps in making better decisions during drug development. However, for AI to be truly effective, it needs access to high-quality data, careful handling of ethical concerns, and a clear understanding of its limitations. In this paper, we also look at the challenges of using AI - database availability, such as lack of transparency in decision making, regulatory frameworks and ways to overcome them by using explainable AI, combining AI with traditional lab research, data acquisition with biobanks and other computer aided drug design software like (autodock, discovery studio, chemdraw, ChEMBL) regulatory engagement, and improving data transparency.
The application of artificial intelligence (AI) in medicinal chemistry has attracted considerable interest in recent years as a possible way to transform the pharmaceutical sector. The discovery of drugs, which involves finding and creating new treatments, is a complicated and lengthy undertaking that often depends on labor-intensive methods like trial-and-error testing and high-throughput screening. Nonetheless, AI methods like machine learning (ML) and natural language processing have the ability to expedite and enhance this procedure by facilitating more precise and effective analysis of extensive data sets. AI-driven techniques have also successfully forecasted the toxicity of potential drug candidates. Various other research initiatives have emphasized the potential of AI to enhance the efficiency and effectiveness of drug discovery procedures. Employing AI in the creation of new bioactive compounds comes with its own challenges and limitations. Ethical factors should be considered, and additional research is necessary to comprehensively grasp the benefits and drawbacks of AI in this field. In spite of these difficulties, AI is anticipated to play a major role in creating new drugs and treatments in the coming years. Generative models can be used to create large libraries of compounds with certain characteristics traits for small molecule drug development projects. In a multiparameter Optimization project, the optimization of an artificial intelligence (AI) molecular generator to explore a given chemical space and propose new well-scored molecules is mostly based on molecular fingerprints and attributes However, the requirement to synthesis the molecules is one of the main obstacles in any computer-aided drug design (CADD) project. Few synthesizability scores have been reported in the literature to be employed in the pipeline of molecular production, while generative models are known to sample a large number of inaccessible compounds. The advancements in high-performance computer hardware and the access to multi-omics data have allowed artificial intelligence (AI) methods to move beyond theoretical research to practical applications across various fields. The effective use of AI methods, especially for analyzing biological data, has drawn the interest of the pharmaceutical sector.
Presently, medicinal chemistry techniques depend to a large extent on hit-and-miss methodology and large-scale testing methodologies [8]. These methodologies consist of screening huge numbers of prospective drug compounds, with the aim of finding those with the intended characteristics. Nonetheless, these methodologies are time-consuming, expensive, and tend to produce results of low accuracy [6]. Besides, they are prone to be limited by the availability of adequate test compounds and the challenge of precisely forecasting their actions in the body [9]. Various AI-based algorithms, such as supervised and unsupervised learning techniques, reinforcement, and evolutionary or rule-based algorithms, have the potential to help solve these issues. These processes are generally data analysis-based on large amounts of data that can be utilized differently [9–11]. For example, the effectiveness and toxicity of novel drug compounds can be forecast using these methods, with more accuracy and effectiveness than through conventional methods [12,13]. In addition, AI-based algorithms can be utilized to find new targets for drug discovery, like the specific proteins or genetic pathways that contribute to diseases [14]. This is able to widen the range of drug discovery beyond the restrictions of more traditional methods and could ultimately result in the discovery of new and more effective medicines [15]. Therefore, while the older methods of pharmaceutical research have been quite successful in the past, they are handicapped by their dependence on trial-and-error experimentation and their failure to be able to predict the behavior of new potential bioactive compounds with any accuracy [16]. AI-based methods, however, can enhance the efficiency and accuracy of drug discovery processes and can result in the creation of more effective drugs.
Another major use of AI in drug discovery is designing new compounds with desired properties and activities. Conventional techniques tend to depend on the discovery and modification of known compounds, which may be a time-consuming and labor-intensive exercise. AI-driven methods can, however, facilitate the quick and efficient design of new compounds with desired properties and activities. For instance, a deep learning (DL) algorithm has been recently trained on a database of known drug compounds and their respective properties, to suggest new therapy molecules [10] with desired features like solubility and activity, proving the prospects of these methodologies in the efficient and fast design of novel drug candidates. DeepMind has recently proved to be a milestone in the domain of AI research along with the emergence of AlphaFold, an unprecedented software platform for the growth of our knowledge of biology [19]. It is a high-performance algorithm that leverages protein sequence information and AI to forecast the proteins' respective three-dimensional structures. This structural biology breakthrough promises to transform personalized medicine and drug discovery. AlphaFold is a huge leap in the application of AI in structural biology and life sciences as a whole. ML methods and molecular dynamics (MD) simulations are now applied in the area of de novo drug design to enhance efficiency and precision. The method of merging these approaches is being investigated to leverage the synergies between them [20]. The application of interpretable machine learning (IML) and DL approaches is also helping towards this goal. By utilizing the strength of AI and MD, scientists are able to more effectively and efficiently design drugs than ever before.
The basic schematics of applying AI techniques to drug discovery and evaluation are summarized in Figure 1. The major procedures include data collection and curation (Figure 1A), compound representation (Figure 1B), and AI methods and their applications in drug discovery (Figure 1C). To provide researchers with a catching-up view of the development in this field, we first introduced representative data resources, molecular representations and descriptors, and AI techniques in drug discovery. Then, we introduced the successful applications of AI to different stages of drug discovery. Finally, we discussed the challenges and future perspectives on applying AI to drug discovery.
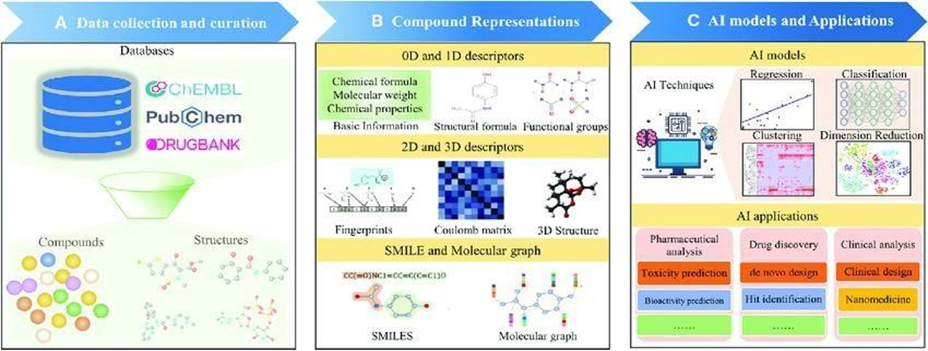
Figure 1: Framework of AI technique application to drug discovery and evaluation.
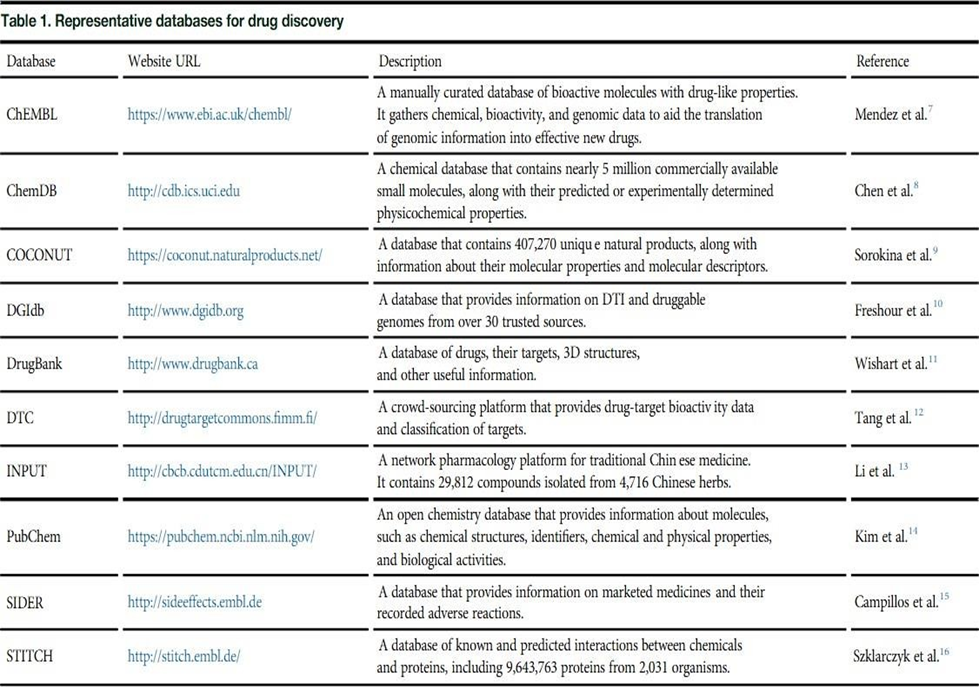
Major procedure includes (A) data collection and curation; (B) compound representations by using molecular descriptors; and (C) AI methods and their applications. ChEMBL is a manually annotated database that presently holds over 2 million compounds with drug-like properties.7ChEMBL collects data on the action mechanisms, molecular properties, absorption, distribution, metabolism, excretion, toxicity, therapeutic indications, and target interactions of the deposited compounds. ChemDB is an open-access database with about 5 million commercially available small molecules and their physicochemical properties, including molecular weight, solubility, and rotatable bonds.8 Besides these, a range of cheminformatics tools like Smi2Depict, MOL pro, AquaSol, and Reaction Predictor are incorporated into ChemDB, thus rendering the database convenient for drug discovery. The Drug-Gene Interaction Database (DGIdb) lists information on drug gene interaction and genes or gene products potentially capable of interaction with drugs.10Up until now, DGIdb encompasses over 40,000 genes and 10,000 drugs associated with greater than100,000 drug- gene interactions. Data are obtained from severalises sources using performance of expert curation and text mining. All of the genes submitted in DGIdb are assigned to 43 categories. Users may either navigate through the genes within a category or type in a list of genes or drugs to retrieve corresponding drug-gene interactions through the search module. Furthermore, DGIdb can be queried programmatically by API via the web-based interface. Drug Bank is an open-access reference drug database.11It now hosts 14,746 drugs with complete details of drug-drug interactions, drug-targets, drug classifications, and drug reactions. Users can search, browse, and extract text, images, and structural data in Drug Bank using the integrated tools. Drug Bank has emerged as the world's most popular resource for drug screening, design, and metabolism prediction. Drug Target Commons (DTC) is an open-access online resource that offers annotated and unannotated drug-target interaction (DTI) data.12For its latest release, DTC adds clinical trial information and disease-gene associations, enabling the chemical biology and drug-repurposing uses of compounds. Being anopen resource, DTC not only offers database dump but also APIto retrieve its deposited data. Discoveries of herb-focused drugs and the scientific explanation of traditional Chinese medicine. PubChem is an open-access chemical information database with the biological, physical, chemical, and toxic data of chemical molecules. All these facts are gathered from over 850 sources. PubChem allows its users to search for chemicals by entering molecular formula, structure, and other identifiers as keywords. Currently, PubChem has developed into one of the primary sources of data for computational drug discovery and design. Side Effect Resource (SIDER) is a drug-side effect database. The SIDER release under current version contains 1,430 drugs, 5,880 side effects, and 140,064 drug-side effect pairs. They can be navigated either through drugs or through side effects. The data have been applied to numerous areas, including predicting drug indications, min-ing side effects, and metabolic dysregulation identification. The Search Tool for Interacting Chemicals (STITCH) is a database that holds known and predicted chemical-protein interactions. The interactions include 9,643,763 proteins of 2,031 organisms, which were gathered from computational prediction, organism-to-organism knowledge transfer, and other data-bases. STITCH can be queried by users in various ways, including chemical and protein names, chemical structures, and protein sequences. For large- scale analysis, data in STITCH can be either bulk downloaded or accessed programmatically by API.
With the explosive growth of natural products, another key point in AI-based drug discovery and analysis is the transfer of molecules into computer-readable format, while keeping their intrinsic physicochemical properties.17Various types of descriptors have been proposed to represent drugs; these descriptors can be classified into four categories in accordance with their dimensionality (Figure 2). To speed up the drug discovery process, a chain of open-source toolkits have been suggested to compute molecular descriptors and structure representations, including OpenBabel18and ChemmineR.19The simplest molecular representation is the zero- dimensional (0D) descriptor; it is derived following the chemical formula ofdrugs.20The 0D descriptor generally encompasses molecular weight, atom number, atom-type count, and other elementary descriptors (e.g., number of heavy atoms).
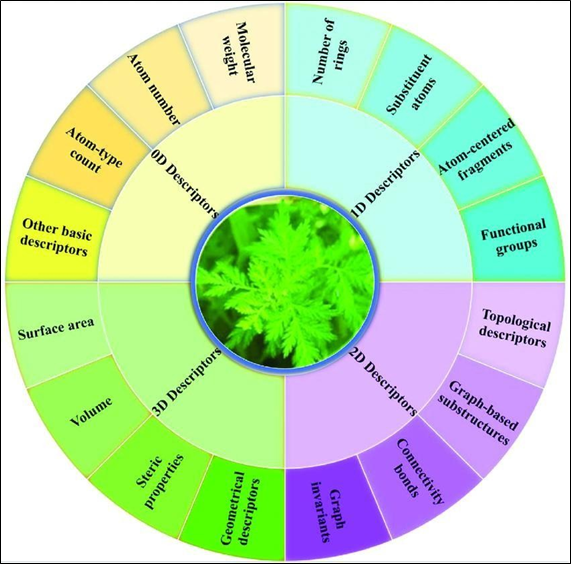
Figure 2. Summary of molecular and structural representation schemes
The 0D descriptor is very straightforward, and it can only retrieve shallow information. The one-dimensional (1D) descriptor maps drugs in accordance with their substructures, including the number of rings, functional groups, substituent atoms, and atom-centered fragments.20The components of the 1D descriptor are usually binary (e.g., 1/0 states the presence/absence of a substituent atom) or the frequency of occurrence of certain substructures. Besides the property- based1D descriptor, simplified molecular-input line-entry system (SMILES)21is another kind of 1D descriptor. SMILES describes drugs as a sequence of characters. SMILES relies upon atom order, and hence, a drug will possess multiple SMILES structures, and the normalization algorithm must be carried out to get canonical SMILES. The two-dimensional (2D) descriptor extends information to the 1D descriptor by taking adjacency, connectivity, and other forms of topological features of atoms into account. Thus, 2D descriptors are usually obtained by encoding a drug as a graph such that in the nodes represent atoms and edges represent bonds. Property-based 2D descriptors often consist of graph invariants, connectivity bonds, graph- based substructures, and topological descriptors. In order to obtain more information, the molecular fingerprint (FP) was suggested for encoding molecules in binary format.22FP represents the presence/absence of certain substructures by a string of a specified length and delineated by 1/0. The standard used2D FPs are molecular access system fingerprints,23daylight-like fingerprint,18and extended-connectivity fingerprints.24The three- dimensional (3D) descriptor represents a molecule in 3D space,25and every atom in a molecule is space-characterized by the x, y, and z coordinates. The 3D descriptor contains spatial and geometrical configuration information; it is of high information con-tent. Therefore, information on surface area, volume, and steric properties can be derived by employing 3D descriptors. Non-property-based3D descriptors, including geometrical fingerprint 26 and pharmacophorefingerprint,27are also present. They are capable of describing advanced physicochemical properties of drugs and are used extensively in drug discovery and virtual screening.

Pharmaceutical analysis consists of the activities of identification, determination, quantification, and purification of raw drug materials; it is a fundamental aspect of drug discovery. Qualitative and quantitative analyses are the two key categories of experimental methods in pharmaceutical analysis. Even though these methods have high accuracy, their price for screening new drug candidates from enormous amounts of natural products remains costly. In contrast with experimental methods, the expenses demanded by computational methods are insignificant. Therefore, AI methods have been employed in pharmaceutical analysis in addition to experimental methods. The exemplary applications of AI methods in pharmaceutical analysis are outlined in Figure 3.
Drug toxicity prediction
Toxicity is a measure of the unwanted or adverse effects of chemi-cals.44Toxicity evaluation is one of the fundamental steps in drug discovery, and it aims to identify substances that have harmful effects on humans.45 However, the in vivo test requires animal tests and thus increases the costs of drug discovery. Computational approaches show the merits of predicting the toxicity of a chemical with low cost and high efficiency.46 As a result, a series of AI method- based approaches have been established to predict the toxicity of chemicals.47,48To evaluate the performance of various computational approaches to predict the toxicity of chemicals, the scientific community proposed the "Toxicology in the 21st Century (Tox21)"challenge.46DeepTox is an ensemble predictive model for the toxicity of chemicals, and its basic framework is a three-layer deep neural network (DNN).49After data cleaning and quality control, the remaining chemicals are represented by applying the above-mentioned 0D to 3D molecular descriptors, which are utilized as input of DNN. Deep Tox pipeline is achieved by hyperparameter tuning and optimizationof a group of hyperparameters, including number of hidden units, learningrate, and dropout rate. Comparative performance on the Tox21 data- set shows that Deep Tox surpasses its counterparts in toxicity prediction.
Drug bioactivity prediction
In real-life, most drugs of natural origin are useless owing to the absence of bioactivity. Therefore, measurement of drug bio activity has been a research field full of energy in drug discovery. Though in vitro and in vivo tests are capable of simulating the activities of molecules in the body, they remain time-consuming and costly. Due to their time economy and cost- effectiveness, AI methods have been successfully used to predict drug bioactivities, including anticancer, antiviral, and antibacterial activities.50–52For instance, Stokes et al. designed a directed message passing neural network capable of predicting antibacterial activity. They first built a molecular graph for each molecule according to its SMILES and then extracted the feature vector from atomic features (e.g., number of bonds by each atom and atomic number) and bond features (e.g., type of bond and stereochemistry).53Using the message passing operation several times, the optimized feature vector was input into the feedforward neural network that produced the antibacterial probability of a molecule.53 Drug physicochemical property prediction Physicochemical drug properties are innate attributes of drugs. Information related to physicochemical properties must be known to understand and simulate drug action. Among the large number of forms of physicochemical properties, solubility is significant due to its effect on pharmacokinetic properties and formulations ofdrugs.54,55 Nonetheless, time-consuming and expensive experiment-ation approaches have hindered the fast prediction of solubility; as such, many efforts have therefore gone into working on AI-assisted solubility prediction models. Panapitiya et al. evaluated various deep learning techniques (i.e., fully connected neural networks, RNNs, graph neural networks, and SchNet) and molecular representation methods (i.e., molecular descriptors, SMILES, molecular graphs, and 3D atomic coordinates) for predicting solubility.54From the same test data, the authors established that the best performance of the fully connected neural network for predicting solubility was achieved using molecular descriptors. Moreover, the authors compared the significance of various features for prediction and concluded that 2D molecular descriptors contributed the most.
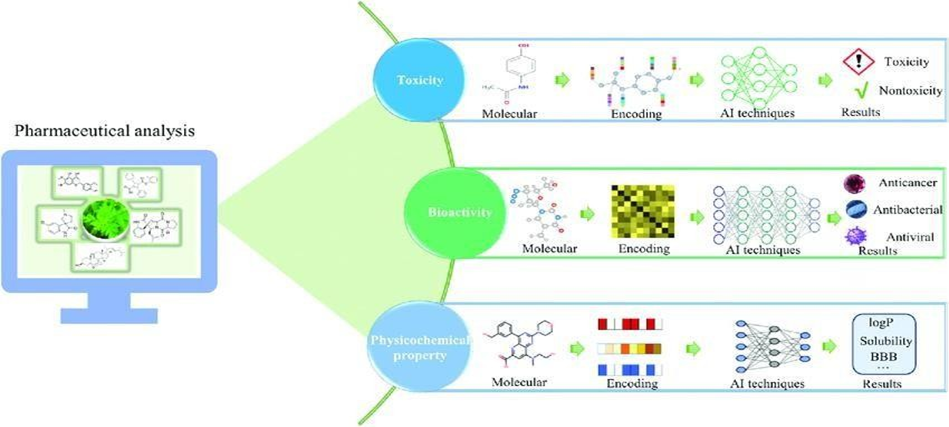
Figure 3. Application of AI techniques to pharmaceutical analysis
AI in natural product-inspired drug discovery
Drug discovery is a method of finding active compounds with therapeutic impacts on the target diseases. While a high-throughput screening method has the capacity to screen thousands of varying com-pounds in individual passes, it remains time-consuming and expensive.56To overcome these issues, AI methods have been utilized to virtually all drug discovery facets. The uses of AI to natural product-inspired drug discovery, including de novo drug design, target structure prediction, DTI prediction, and drug-target binding affinity prediction
De novo drug design
De novo drug design is a process of producing new drug-like molecules without an initial template. While traditional structure-based and ligand-based drug design approaches have improved the discovery of small-molecule drug candidates, they respectively depend on understanding the active site of a biological target or pharmacophores of a known active binder,57preventing their applications to contemporary drug discovery. The AI techniques boom has brought new chances to de novo drug design and promoted the drug discovery process. Over the past few years, many deep learning-based models have been proposed for de novo drug design, including the reinforcement learning-based model ReLeaSE,58the encoder- decoder-based modelChemVAE,59the GAN-based model GraphINVENT,60and the RNN- based model MolRNN.61Another important aspect of de novo drug design is molecular representation. SMILES, fingerprint, molecular graph, and 3D geometry have been utilized as input of deep learning algorithms. The basic framework of deep learning-based de novo drug design approaches is depicted in the left upper corner of
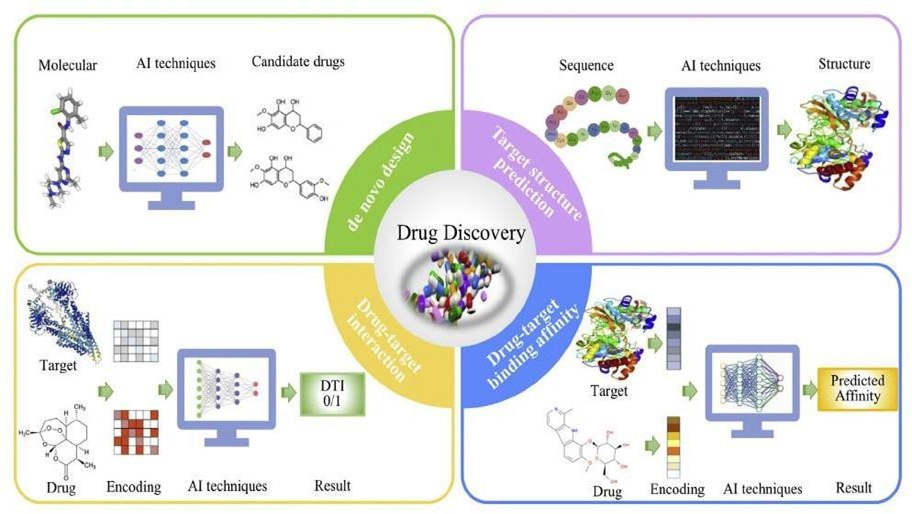
Figure 4. Detailed information about deep learning-based de novodrug design models is provided recent reviews.
Target structure prediction
The majority of drug targets are proteins with significant functions in enzymatic processes, cell signaling, and cell-cell transduction. Protein functions are defined by structures. Though traditional experimental methods, including X-ray crystallography, cryogenic electron microscopy, and nuclear magnetic resonance spectroscopy, have been suggested to find protein structures, they remain time-consuming and expensive.63As mentioned, experimental methods have only deciphered the structures of100,000 distinct proteins, which represent a minor fraction of known proteins.64Thus, the creation of new methods to bridge the gap between the number of protein sequences and known protein structures is an imperative requirement.65With the accelerated increase in computational power and the breakthroughs of AI methods, numerous computational methods have been suggested for protein structure prediction. The schematics of the basic schematics of computational protein structure prediction models are shown in the right top corner of Figure 4. The best-performing method is the neural network-based Alpha Fold approach developed by DeepMind, and it can predict the 3D structure of proteins from their amino acid sequences and achieve experimentally competitive accuracies. The algorithm and architecture description of Alpha Fold is given in Senior et al.66The source code of Alpha Fold is hosted at https://github.com/deepmind/alphafold.
DTI prediction
DTI prediction is the interaction between protein targets and chemical compounds in living organisms.67 DTI prediction is a crucial process in drug discovery. Experimental methods have, therefore, been employed to predict DTI, including co-immunoprecipitation,68 phage display technology,69 and yeast two-hybrid.70 The wet laboratory methods are, however, time- consuming when employed to predict DTI. In recent times, the constantly growing biological data have laid the ground for in silico prediction of DTI. As such, computational techniques are increasingly being employed in the prediction of DTI. These techniques, which were discussed in a recent review,71 can be divided into the following categories: ligand-based techniques, docking simulations, gene ontology-based techniques, text mining-based techniques, and network-based techniques. Deep learning-based techniques, relative to other types of techniques more often show improved performance in DTI prediction.72 The typical flow of the DTI prediction procedure based on deep learning is indicated at the lower left corner in Figure 4. Compounds and proteins are first encoded utilizing their respective features. Subsequently, the feature embedding of proteins and compounds serve as the inputs of deep learning techniques. Corresponding to this approach, the models based on deep belief neural network,73 CNN,72 and multiple layer perceptron74 have been suggested for drug-protein interaction prediction, significantly advancing drug discovery. In real life, most diseases don't have clearly defined targets. Therefore, discovering drugs for these diseases is not feasible using the above methods. Zhu et al. recently suggested a deep learning-based efficacy drug candidate predicting system (DLEPS) capable of picking out drug candidates based on the shifts of gene expression profiles instead of specific targets.75 Initially, the compounds were SMILES encoded and employed as CNN input to regress gene expression alterations. Afterwards, the compound effectiveness against diseases was assessed based on gene signatures defining particular diseases and ranked with an approach similar to gene set enrichment analysis. DLEPS offers new information for the identification of new drugs for complicated diseases.
Drug-target binding affinity prediction
DTI prediction is, in most instances, considered a binary classification problem, but binding affinity between drug and target is not considered.67 Binding affinity indicates the intensity of drug-target pair interactions, and it is significantly informative for drug discovery. Though binding affinity can be experimentally measured by determining dissociation and inhibition constants, the time cost and economic expenses of these processes are very high. Hence, computational methods for the prediction of binding affinity is required. In 2018, Öztürk et al. developed the first deep learning model, named DeepDTA, for the prediction of binding affinity between drugs and their targets.76 DeepDTA employed the drug and the target by encoding them using SMILES and amino acid letters, respectively, and they were utilized as input to CNN. The general framework of DeepDTA is presented at the right bottom corner of Figure 4. The comparative results illustrated that DeepDTA dominated KronRLS77 and SimBoost78 for drug-target binding affinity prediction. Inpired by DeepDTA, a series of deep learning-based models has been sequentially introduced, including WideDTA76 and DeepAffinity,79 that have become effective tools in drug discovery
Advanced uses of AI in drug design
AI in prediction of drug synergism/antagonism
Synergism and antagonism are the two types of drug combination effects. The former can overcome primary and secondary drug resistance, and it is useful for the treatment of cancers,80 AIDS,81 and bacterial infections,82 while the latter decreases the efficacy of drugs. With the constantly growing number of drugs, their potential combinations are astronomical. Therefore, experimentally exploring drug combination effect is time-consuming and expensive. The developments of AI methods have rendered them suitable for investigating potential drug combinations at reduced cost and with greater efficiency. Li et al. In 2015 suggested a Bayesian network model for investigating and examining drug combinations.83 Wildenhain et al. in the same year developed a random forest-based model to predict compound synergism from chemical-genetic interactions.84 Preuer et al. recently suggested DeepSynergy,85 a deep learning-based model for anticancer drug synergism. The chemical data of drugs and genomic data of diseases were the inputs for DeepSynergy, which propagated them through the network to the output unit. The comparative performance on a publicly available dataset of synergy showed that DeepSynergy performed better than its competitors in predicting drug synergism. The web server and source code of DeepSynergy are available at www.bioinf.jku.at/software/DeepSynergy and https://github.com/KristinaPreu er/DeepSynergy, respectively.
AI in nanomedicine design
Nanotechnology has been used to design nanomedicines through the application of nanometric- scale materials in the clinical environment.86 Nanomedicines are created by nanometric-scale materials, and therefore, they can cross the barriers to engage with targets in the body. Currently, there are some nanomedicines that have been approved by the U.S. Food and Drug Administration, and they have performed better in the treatment of cancers87 and HIV-1 infection.88 Yet, the absence of quantitative and qualitative knowledge of nanomaterial properties and biological responses hindered the extensive use of nanomedicines. Nanotechnology in combination with AI offers new solutions to resolve this issue. For instance, Li et al. suggested an ANN for nanomedicine composition optimization.89 Muñiz Castro et al. created a pipeline for formulating nanomaterials through 3D printing can forecast the nanomaterial's extrusion temperature, filament mechanical properties, and dissolution time.90 Besides, the effectiveness of a nanomedicine is also influenced by cellular uptake. Thus, a prediction model for cellular uptake will greatly assist researchers in forecasting nanomedicine effectiveness. Based on an ANN, Alafeef et al. created a platform for forecasting nanoparticle cellular internalization in various cell types.91 Other applications of AI in nanomedicine design and their principles were compiled in a recent exhaustive review.80
AI in oligonucleotide design
In addition to drugs based on natural products, oligonucleotide therapeutics made up of short DNA or RNA strands have emerged as a new class of drugs.92 Antisense oligonucleotides (ASO), small interfering RNA (siRNA), and CRISPR-Cas (clustered regularly interspaced short palindromic repeats)-associated protein are the primary oligonucleotide therapeutics systems that make possible the specific treatment of wide-ranging diseases. Because experimental designing these oligonucleotides will be very costly, the AI methodologies have also been employed to assist researchers to identify and design the oligonucleotide-based drugs. For instance, Chiba et al. suggested a machine learning-based model, eSkip-Finder, to find effective exon skipping ASOs.93 Dar et al. designed SMEpred to predict the efficacy of siRNAs.94
Molecular Docking
Molecular docking is a key drug discovery tool used to assist scientists in knowing how small molecules (such as drugs) bind to biological targets (most often proteins). Classical molecular docking had the researcher using trial-and-error experimental techniques, heavily dependent on scientific expertise and laboratory work. Nevertheless, with the arrival of artificial intelligence (AI), molecular docking has become faster, more accurate, and less costly. This paper discusses how AI and molecular docking intersect, their advantages and drawbacks, as well as what they are contributing to the drug discovery of the future. Before diving into AI’s role in molecular docking, it’s essential to understand what molecular docking entails. In essence, molecular docking is a computational method used to predict the preferred orientation and binding affinity of a ligand (a molecule that binds to a target, such as a drug or peptide) when it interacts with a receptor (usually a protein). The objective of docking is to locate the most energetically favored binding mode of the ligand-receptor complex, which is important for finding potential drug candidates. Classically, molecular docking involves solving intricate equations representing the intermolecular interactions. The solution returns a researcher a ranking of the potential modes of binding using computed energy scores. The predictions have the capability of guiding experimental studies by scheduling the molecules to be tested secondly, hence accelerating the process of drug discovery.
The Application of AI in Molecular Docking
Artificial intelligence, more specifically machine learning (ML) and deep learning (DL), has come to assume a revolutionary role in molecular docking. This is how:
Conventional molecular docking approaches are based on pre-established algorithms and heuristics to model molecular interactions. Although these models can be useful, they tend to be limited in accuracy and computational efficiency. AI methods, especially deep learning, have the potential to improve the accuracy of docking predictions by learning from enormous amounts of data, such as the physical and chemical properties of molecules and their interactions. For instance, AI algorithms can be taught to recognize known molecular interactions, so they may predict the binding affinity of novel molecules with increased accuracy. Machine learning models may learn complex patterns in molecular binding, challenging traditional approaches, by learning from large sets of protein-ligand complexes and training models on them.
AI can greatly accelerate the process of drug discovery, particularly in the initial stages where thousands of candidate drugs have to be screened. Conventional docking processes may take days or weeks to process large datasets, whereas AI-based docking is capable of processing these within a small fraction of the time. By using high-throughput screening, AI is able to rapidly forecast which compounds are likely to bind well to a target protein, shortlisting candidates for further testing. The speed-up enables researchers to better prioritize drug candidates, saving time and money in the hunt for good compounds. In a business where time is as important as it is, having the capacity to carry out faster screening may be a game-saver.
Virtual screening is a computer method for selecting potential drug candidates by modeling the way they may bind to a target protein. With AI, virtual screening becomes much more accurate and efficient. Deep learning algorithms can examine the three-dimensional shape of proteins and ligands with great accuracy and predict binding interactions in a manner that conventional algorithms frequently cannot. By merging AI with virtual screening, scientists can probe more extensive libraries of chemicals and find new drug candidates that would have otherwise been overlooked by traditional means. AI models can even make predictions regarding how slight chemical adjustments to a ligand may enhance its binding affinity, allowing for rational drug design and optimization.
One of the most important challenges in drug discovery is the design of molecules that bind specifically and efficiently to a target protein without inducing off-target effects. AI can optimize drug-target interactions by gaining deeper insights into molecular docking. With reinforcement learning and generative models, AI can suggest new chemical structures or modifications to enhance the binding affinity and selectivity of drug candidates. Through the integration of AI-optimized optimization, scientists can select the best drug candidates sooner, improving the chances of clinical trial success. AI can also help to create drugs with fewer side effects by predicting potential off-target interactions, a significant barrier to drug development.
Limitations and Challenges of AI in Molecular Docking
Though full of promise, AI in molecular docking is not without limitations:
Large amounts of data are needed to train AI models effectively. The quality and availability of the data in terms of experimentally validated protein-ligand complexes are limited. Large- scale databases such as the Protein Data Bank (PDB) and PubChem are available but may not include all the possible interactions or have high-quality data for every protein of interest. In addition, AI models might have issues with noisy and incomplete data, potentially resulting in wrong predictions.
Deep learning models, though powerful, are sometimes referred to as "black boxes," that is, their decision-making process is not necessarily transparent. This lack of explainability can become a major impediment to their use in drug discovery, particularly in an industry where knowing the rationale for predictions is essential. Techniques such as attempting to make AI models explainable are being pursued, but this remains a work in progress.
Incorporating AI-based molecular docking into current drug discovery pipelines can be challenging. Pharmaceutical firms tend to use conventional techniques and tools, and embracing AI involves heavy infrastructure, computational power, and training. But as AI technology matures and its advantages become clearer, its incorporation into drug discovery pipelines will increasingly become seamless.
Molecular Docking and Structure-Based Drug Design Studies
Molecular docking is a well-established and widely used methodology in drug design. A substantial number of studies are available in which a diverse array of approaches has been applied for the discovery of novel bioactive molecules. Recent cases involving different docking strategies combined with other molecular modeling methods are examined next.
5.1. Discovery of Mycobacterium tuberculosis InhA Inhibitors Using SBVS and Pharmacophore Modeling
Trans-enoyl-ACP reductases are NADH-dependent enzymes involved in fatty acid biosynthesis. The enzyme from Mycobacterium tuberculosis (InhA) promotes the synthesis of long-chain fatty acids, namely mycolic acids, which are an essential component of the bacterial cell wall [205]. An important enzyme in tuberculosis drug discovery, InhA is the molecular target for the tuberculostatic drug isoniazid [206]. The compound is a pro-drug that loses its hydrazine group as it reacts with the enzyme cofactor NADH, forming an isonicotinic-acyl- NADH complex that inhibits the catalytic activity of InhA (Figure 6).

Figure 5. (A) Structure of the tuberculostatic drug isoniazid; (B) The isonicotinic-acyl moiety covalently bound to NADH in the binding site of InhA (PDB 1ZID, 2.70 Å). The protein backbone is represented as a cartoon. The isonicotinic-acyl fragment (carbon in yellow) and NADH (carbon in white) are shown as sticks.
The Future of AI in Molecular Docking
The future of AI in molecular docking is full of interesting potential. With increasingly advanced AI models and increasingly high-quality data, molecular docking will be increasingly accurate, efficient, and a part of the drug discovery process. Even the integration of AI with other new technologies, such as quantum computing, might further speed the drug discovery process and allow for more personalized, effective treatments. In summary, AI is more than a tool; it is a revolutionary driver in the pharmaceutical sector. By optimizing molecular docking, AI is assisting scientists to find drug candidates quicker, better optimize interactions, and save money and time during the drug discovery process. With further development, the technology is poised to change the future of drug discovery, bringing new treatments to market quicker and enhancing world healthcare outcomes.
REFERENCES

Uttam Kumar*, Priyal Jain, Dr. Jitendra Banweer, Artificial Intelligence Based Drug Designing, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 5, 1535-1552. https://doi.org/10.5281/zenodo.15380466
 10.5281/zenodo.15380466
10.5281/zenodo.15380466