
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Chemistry, College of Pharmaceutical Sciences Govt. Medical College, Thiruvananthapuram, 695011
Breast cancer is a disease in which cells in the breast grow out of control.It is the most frequently diagnosed cancer in women.Various enzymes and receptors are involved in the development and progression of breast cancer.That is,development of breast cancer is a multi-step process that involves several cell types, and it is still challenging to prevent completely.Now a days, breast cancer detection and diagnosis have become much more accurate.A correct timely diagnosis and appropriate treatment plan can achieve the predictable therapeutic result. There are various heterocyclic ring systems which act on diverse biological targets. Benzimidazole is such a system which consist of an imidazole ring attached to the benzene nucleus. The pharmacological potential of the system on diverse targets are studied so far. It is commonly recognised that benzimidazole and its derivatives have a significant role in the creation of new anticancer medications.The anticancer efficacy of several benzimidazole derivatives towards different receptors, such as EGFR, VEGFR, Topoisomerases, Aromatase, Peptidyl-prolyl isomerase (PIN1) enzyme, cMet kinase, Poly ADP-ribose polymerase, etc., has been investigated.This review article focuses on the anti-breast cancer effects of benzimidazole derivatives through targeting various receptors.
CANCER
Cancer is a highly invasive disease characterised by the genetic or epigenetic alterations in somatic cells which result in uncontrolled cell growth and dissemination of abnormal cells.Depending on the kind of tissue that the cancer was found in, there are over 200 different types of cancers[1]. Metastases is the major cause of death due to cancer. Tobacco use, alcohol intake, unhealthy diet, less physical activity, pollution are some of the risk factors for development of cancer. As per WHO report, cancer caused nearly 10 million deaths globally and an estimated 19.3 million new cases in 2020[2]. Lung, prostate and colorectal cancer is most common in males, while breast cancer is most frequent in women followed by colorectal, lung and cervical cancer[3]. Now a days, most frequently occurring cancer is the breast cancer and it is the leading cause of death among women. Genetic causes (BRCA 1 and BRCA 2 are abnormal genes, the inheritance of the same may increase the risk of breast cancer), alterations in hormonal levels, sedentary life style(high fat intake, obesity, consumption of alcohol etc.), environmental causes like radiation etc. are some of the risk factors to breast cancer[4].If one first-degree relative has the disease, the chance of acquiring breast cancer is around two times higher, and it may be five times higher if the relative had breast cancer while they were younger[5].Breast cancers that are sporadic, meaning they lack a significant genetic component, typically manifest later in life, possibly owing to postmenopausal breast cancers[6].In contrast, breast cancers that are inherited tend to develop earlier in life, which is indicative of a genetic predisposition[7].
Subtypes of breast cancer:
[8] there are mainly 4 molecular subtypes of breast cancers based on the receptors involved;
these tumours are estrogen receptor positive and/or progesterone receptor positive, but human epidermal growth factor receptor negative (ER +ve and/or PR +ve but HER2 -ve). These types of tumours grow slowly. Itis connected with a favourable prognosis and accounts for around 60% of breast cancer[9].
these tumours are positive for both estrogen receptor and human epidermal growth factor receptor (HER2) but negative for progesterone receptor. It represents 30% of breast cancer and is associated with poor prognosis.
these tumours are positive for HER2 but negative for both ER and PR. These grow faster than the luminal tumours, it accounts for 10% of breast cancer and associated with poor prognosis[10]. It can be treated with targeted therapy towards HER2.
these tumours are negative for ER, PR and HER2. It occurs due to the mutation of BRCA1 gene. Constitutes 15–20% of BC. It is linked with higher aggressiveness and a poorer prognosis when compared to other BC subtypes, and often affects younger women[11]. BRCA is an acronym for “BReastCAncer gene”. There are two types of BRCA genes: BRCA1 and BRCA2. The proteins that the BRCA1 and BRCA2 genes encode are essential for preserving genomic stability and DNA integrity. People with a mutation in one of these genes have an increased risk of developing breast cancer, and are more likely to have triple-negative breast cancer (TNBC)[12].
Stages of breast cancer:
Tumour size, nodal involvement, metastasis presence, and some biomarkers such as progesterone, estrogen and HER2 receptors are used to determine the stage of breast cancer. Stage 0, non-invasive breast cancer is also called as Ductal carcinoma in situ (DCIS).Stages I, IIa, and IIb are associated with early invasive cancer, while stages IIIa, IIIb, and IIIc are associated with locally advanced cases.Breast cancer in all of these stages is not metastatic. Stage Four breast cancer is metastatic[13].
Stage 0, non-invasive breast cancer:
Most frequently detected by mammography, which only shows microcalcifications in the breast duct. Up to 40% of DCIS cases will develop to invasive breast cancer if treatment is not received[14].Treatment options for DCIS include radiation therapyand lumpectomy or with mastectomy. Patients withoestrogen receptor-positive DCIS should undergo endocrine therapy for five years[15].
Stage I – III Early Invasive and Locally Advanced, Nonmetastatic breast cancer:
Surgery, and radiation along with preoperative and postoperative systemic therapies, such as chemotherapy, endocrine therapy, immunotherapyare used to treat nonmetastatic breast cancer.
Stage IV metastatic breast cancer:
Metastatic breast cancer is rarely cured and the main objectives of treatment are to reduce symptoms, prolong life, and maintain quality of life.As needed to give palliation, symptomatic therapies have to be suggested[16]. A correct timely diagnosis and appropriate treatment plan can achieve the predictable therapeutic result. The treatment usually includes surgery, chemotherapy and radiotherapy. Hormonal therapy has recently been added to the main procedure for the treatment of breast cancer. Resistance and serious adverse effects to the existing medications are a problem in cancer chemotherapy. Thus, targeted therapy is a convenient method to minimize this problem. Estrogenreceptors[17] and Human Epidermal Growth Factor receptor 2[18] are the primary targets that are taken into consideration for chemotherapeutic development. Novel chemical classes known as AIs(aromatase inhibitors)[19], SERM(selective estrogen receptor modulators)[20], and SERDs(selective estrogen receptor down-regulators)[21] are utilised against these targets.Epidermal growth factor receptor[22], heat shock protein[23], poly-(ADP ribose) polymerase[24], and vascular endothelial growth factor receptor[25] are the major molecular targets for treatment of TNBC[1].
BENZIMIDAZOLE
Several five or six membered aromatic systems with heteroatoms are highly researched due to their interesting pharmacological profile. The heterocyclic systems like thiadiazole, oxazole, quinazolines, thiophenes etc. are having various pharmacological activities like anticancer, anti-inflammatory, antiulcer, antimicrobial etc. Benzimidazole is the benzo derivative of imidazole, a class of heterocyclic aromatic bicyclic compound, containing a benzene ring attached to the 4, 5- positions of imidazole ring. It was first prepared by Hoebrecker in 1872 by reducing 2-nitro-4-methyl acetanilide.
Nitrogen heterocycles exhibit various pharmacological activities due to resemblance with many natural and synthetic molecules[26].
Two hetero-nitrogen atoms and an electron-rich aromatic system allow the characteristic fused benzene and imidazole rings to interact non-covalently with a variety of biological targets, which is believed to be the cause of the wide range of pharmacological activities of benzimidazole-containing agents[27,28]. Its various biological activities like antifungal[29], antibacterial[30], anticancer[31], anti-inflammatory[32], antihypertensives[33], antiviral[34] etc. are studied. Due to its diverse biological profile and synthetic uses in medicinal chemistry, the benzimidazole nucleus is sometimes referred to as the ‘‘Master Key’’[1].
Table 1: various benzimidazole ring system containing drugs available in the market
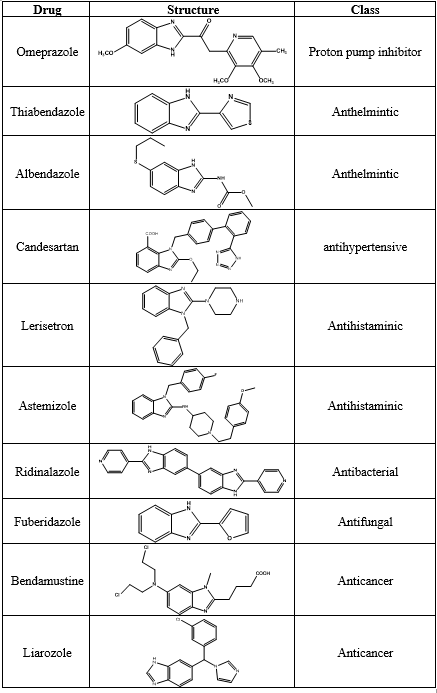
The anticancer efficacy of several benzimidazole derivatives towards different receptors, such as EGFR[35], VEGFR[36], Topoisomerases[37], Aromatase[38], Peptidyl-prolyl isomerase (PIN1) enzyme[39], cMet kinase[40], Poly ADP-ribose polymerase[41], etc., has been investigated.This review focuses on the anti-breast cancer activity of various benzimidazole derivatives by the action on diverse receptors.
ESTROGEN RECEPTOR
Estrogen plays a key role in mediating the maturation, proliferation, differentiation, apoptosis, inflammation, metabolism, homeostasis and brain function and influences the growth and development of breast cancer[42].The nuclear receptors ER? and ER? are activated through the binding of estrogens. The expression levels of these receptors vary among tissues. The ER? plays prominent role in mammary gland and uterus and the over expression of the same is associated with breast cancer physiology[43]. The ER? counteracts the Er? promoted cell hyperproliferation. A logical approach for the treatment of estrogen-sensitive breast cancer is the use of anti-estrogens that inhibit the estrogen functioning in breast cancer cells and these are called selective estrogen receptor modulators (SERMs). Fikriye Zengin Karadayiet. al., in 2020[43] synthesized novel ethylsulfonyl indole-benzimidazole derivatives by substituting the first(R1) and fifth(R2) positions of benzimidazole and indole groups respectively. Subsequently characterised through 1H NMR, 13C NMR, Mass spectroscopy, performed the in-silico docking analysis and screened the anticancer activity.
Cytotoxic assays on human cancer cell lines: MTT assay was used to measure cell viability of MCF-7, MDA-MB-231 and HEPG2 cell lines. To screen large numbers of derivatives before pursuing selected compounds in more detail, cytotoxicity of all ethylsulfonyl derivatives were studied at four doses, 0.25µM, 2µM, 16µM and 40µM in MCF-7, an ER+ and TP53 wild-type breast cancer cell line. There was significant interaction between R1 and R2 groups based on the cell viability scores, which showed that the substitution on indoles could modify the activity of benzimidazoles differentially. The p-fluorobenzyl R1 group is one of the most effective R1 moiety outstanding from the rest of substitutions.
Upon analysis, for further screening they selected several compounds that were highly effective in reducing the viability at the highest dose 40µM: Compounds 1a(%relative viability= 24.36%), 1b(40.38%), 1c(44.16%), 1d(26.90%), 1e(32.85%), 1f(43.76%), 1g(42.19%), 1h(39.58%), 1i(59.55%), 1j(52.00%), 1k(40.52%), 1l(45.69%), 1m(33.53%)
Among these 1i-1l spanning the full p-fluorobenzyl series exhibited similar activity at 40µM whereas 1k, 1l and 1m were also significantly antiproliferative at a relatively lower concentration of 16µM.
Table 2:Chemical structures of selected derivatives
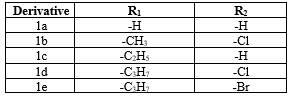
The selected compounds where then screened on different cell lines, showed that the effect on estrogen responsive cell lines MCF-7 and HEPG2 where more similar to each other than ER negative MDA-MB-231 cells at lower concentrations. The compound1m exhibited low IC50 values for TP53 wildtype MCF-7 and HEPG2 cells (19.23µM and 24.10µM respectively) while it was not as effective in MDA-MB-231, a cell line with mutant TP53 allele. The derivatives showing higher potencies have also taken for docking analyses on the ER? ligand binding domain (PDB ID: 1a52). Considering the docking results, one possible reason of the higher activity shown by compounds 1l and 1m could be the increased amount of halogen bond interactions. Also, the bromine group may enhance lipophilic characteristic of indole moiety creating a more successful binding pattern. In-silico findings and literature reviews suggested that ER? is the most favourable indole-benzimidazole target in comparison to Er?, tubulin and VEGFR. The summary of structural activity relationships concluded in this study is the indole-benzimidazoles that have either p-fluorobenzyl, difluorobenzyl or small alkyl group at R1 in addition to electron withdrawing group at R2 may result in relatively more effective anticancer activities.
AROMATASE ENZYME
Estrogen has the ability to influence the development as well as the growth of breast cancer[45].Aromatase enzyme is sometimes referred to as estrogen synthetase or estrogensynthase[46].It catalyses the last and rate-limiting step in the biosynthesis of estrogen[47].Aromatase catalyse the conversion of androstenedione (ASD),testosterone (TST), and 16?-hydroxytestosterone to estrone (E1), estradiol (E2), and 17?,16?-estriol (E3), respectively. It is highly specific in activity since it is the only enzyme in humans that can catalyse the conversion of androgens to estrogens[48]. Breast cancer (BC), which is mostly estrogen- or progesterone-dependent, is a fatal kind of cancer that primarily impacts postmenopausal women[49-51].Premenopausal women generate the majority of their estrogen in the ovaries, whereas postmenopausal women produce the majority of their estrogen in the body's peripheral tissues[52]. Thus, aging is one of the most important risk factors of breast cancer [53]. Aromatase Inhibitors are utilised to either stop the synthesis of oestrogen or stop oestrogen from acting on receptors[54].It has been observed that BC responds to the generation of estrogen at the cancerous site, such as the breast's adipose tissue, which can be blocked by Aromatase Inhibitors[55].
Azizah M Malebari[56]et.al., in 2023 synthesised and evaluated the benzimidazole derivatives of the natural product vanillin as a novel aromatase inhibitor. Using the MTT method, the cytotoxicity of vanillin-benzimidazole analogues (3a-3f) was assessed against five typical adenocarcinomas: breast (MCF-7, MDA-MB-231), ovarian (SKOV3), liver (HepG2), and colon (HCT-116) cancer cells. Doxorubicin was used as a reference molecule.Compounds 3a-3c outperformed vanillin in terms of their moderate to highly effective antiproliferative activities towards the majority of the examined cancer cell lines. The most effective of these was3a, 4-fluorophenyl substituted benzimidazole (IC50 values in MCF-7, MDA-MB-231, and SKOV3 were 0.6, 1.01 and 5.37µM, respectively). Replacement of fluorine substituent with chlorine atom 3b somewhat reduced the activity in MCF-7 and MDA-MB-231 (IC50 values 1.33 and 2.36µM respectively) and diminished the activity in SKOV3 with IC50 39.5µM. addition of chlorine atom at the para-position of phenyl ring to compound 3b to get 2,4-dichlorophenyl benzimidazole 3c preserved the compound's well-to-moderate antiproliferative activity in MCF-7 and MDA-MB231 with an IC50 value of 4.89 and 8.65µM respectively.Replacement of the 4-fluoro substituent with bulkier group 4-sulfamoyl, 3d diminished the activity and the introduction of heterocyclic substituents at the benzimidazole moiety as morpholine 3e and pyrrolidine 3f, resulted in a marked decrease in activity in all cancer cell lines.
Ramit Singla et. al., in 2017[44]combined indole nucleus with benzimidazole to create novel indole-benzimidazole hybrids. Analysed the anti-proliferative activity of all derivatives (N-benzylated indole-benzimidazole derivatives and N-H indole-benzimidazole derivatives) using ER? responsive T47D breast cancer cell lines and ER? binding assay. The bromo substituted derivatives, compounds 2a(N- benzylated indole-benzimidazole derivative) and 2b(N-H indole-benzimidazole derivative)found to be more active with IC50value of anti-proliferative activity15.48 ± 0.10µM and 4.99 ± 0.6µM respectively,hence were taken for ER? targeting gene expression investigations.Experiments using Western blotting and RT-PCR confirmed that both substances altered the expression of mRNA and receptor protein of ER?, halting the subsequent transactivation and signalling cascade in T47D cell lines.According to structural analysis based on an induced fit simulation research, compounds 2a and 2b bind in an antagonistic conformation via strong hydrogen bonds and Van der Waals forces, much like bazedoxifene (standard drug).All of these findings clearly showed that compounds 2a and2b have new, robust ER? antagonist qualities and will be useful in the search for SERM that may be used to treat breast cancer.
Table 3: substitutions in selected derivatives
The fluorimetric aromatase (CYP19A) assay kit was used for the screeining of in vitro aromatase inhibitory activity of most potent antiproliferative compounds (3a-3c). Letrozole was used as the reference standard. Compound 3a exhibited the most promising aromatase inhibition, with an IC50 of 0.064 µM, comparable to letrozole, while compound 3b and 3c inhibited aromatase with IC50¬ 1.16 and 2.87µM respectively.
Table 4: Aromatase Inhibitory Activity Of Most Promising Compounds
In the docking investigation, these substances interacted well with the aromatase enzyme's active sites and also shown favourable pharmacokinetic characteristics.To sum up, the 4-fluorophenyl substituted benzimidazole derivative, compound 3a showed promise as an aromatase inhibitor for the treatment of breast cancer.
U V Cevik et. al.,in 2020[38] synthesized sixteenbenzimidazole-triazolothiadiazine derivatives and studied their aromatase inhibitory activity.
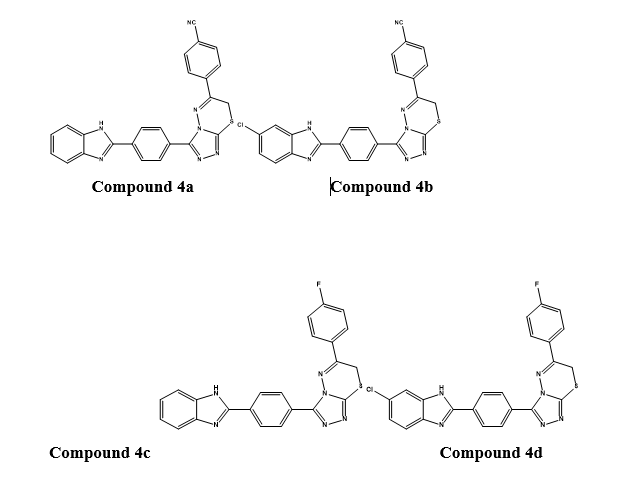
Initially, analysed the in vitro anticancer potential against MCF-7 breast cancer cell lines by MTT assay by taking cisplatin as reference standard.The most potent compounds were the 4-cyano derivatives 4a and 4b with IC50value of 0.016 ± 0.001 and 0.018 ± 0.001 µM, respectively compared to IC50 = 0.020 ± 0.009 µM for the reference drug cisplatin.Furthermore, the most promising activity was observed for the compounds 4c and 4d bearing 4-fluorophenyl derivatives with IC50 values 0.119 ± 0.005 and 0.110 ± 0.005 µM, respectively compared to the reference drug cisplatin(IC50 value 0.020 ± 0.009 µM).
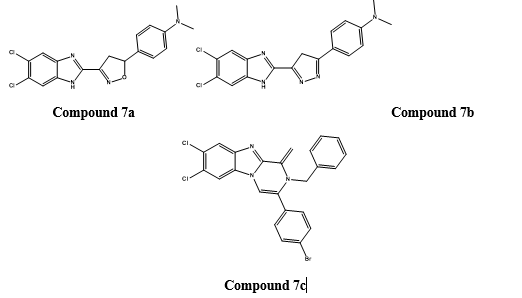
The in vitro anti-aromatase activity of the selected compounds was evaluated using Aromatase-CYP19A Inhibitor Screening kit, using letrozole as reference drug. The best result was shown by the compound 4a with IC50 value of 0.032 ± 0.001 µM (letrozole = 0.024 ± 0.001). Then the docking study of the compound 4a was carried out on the crystal structure of human aromatase enzyme. It was found that two ?–? interactions were formed by the benzimidazole ring in the structure with Arg115 and Phe134.Hydrogen bonding is formed between the hydroxyl of Ser314 and the nitrogen atom of the cyano- group at the C-4 position of the phenyl ring. This interaction is believed to be crucial for the inhibitory activity.
HER 2 AND EGFR RECEPTORS
Tyrosine kinases are a family of enzymes, which catalyse the phosphorylation of tyrosine residues using ATP. The receptor tyrosine kinase family include four members: EGFR (ErbB1, HER1), ErbB2 (HER2), ErbB3 (HER3), ErbB4 (HER4). These kinases play a vital role in the cell growth and metabolism. The up-regulation of these receptors results in various types of malignancies like breast, colon, prostate and lung cancer. Human Epidermal Growth Factor Receptor plays a key role in development of breast cancer. Normally, HER2 receptors help control how a healthy breast cell grows, divides, and repairs itself. Breast cancers with HER2 protein over expression are called HER 2 positive, and which grow faster [57]. Thus, HER2 targeting is a convenient way for the treatment of HER2 positive BC. Over 50% of cases with triple-negative breast cancer (TNBC), which is defined by overexpression of EGFR in addition to the absence of ER, PR expression, and HER2 amplification. Thus, one potential treatment for these malignant tumours could be to target the EGFR pathway[58].
B Chu et.al.[59]in 2015 studied the antitumour activity of 2-aryl benzimidazole compound (2-chloro-N-(2-p-tolyl-1H-benzo[d]imidazol-5-yl)acetamide (5a) on EGFR and HER2.
The anticancer effects of 5a on human breast cancer cells were evaluated in vitro using a group of nine breast cancer cell lines. These cell lines expressed varying levels of EGFR and HER2, including EGFR-positive, HER2-positive and EGFR/HER2- double-negative cells. MTT results showed that, human breast cancer cell lines containing EGFR gene amplification with high expression of EGFR (MDA-MB-468) showed an IC50 value of 3.3?M, whereas the other cell lines with EGFR gene amplification but with low EGFR expression, such as HCC1937, showed an IC50 of 9.02?M. Also, breast cancer cells that overexpress HER2 exhibited a similar response to 5a. The IC50 value from a cell line with high expression of HER2 (BT-474) was 3.58?M, whereas the other cell lines with lower levels of HER2 expression, such as MDA-MB-453, showed an IC50 equal to 4.91?M. The results suggest that cell lines with EGFR and HER2 gene amplification are more susceptible to 5a.
POLY-ADP RIBOSE POLYMERASE (PARP)
For cells to survive, DNA repair is essential.A very precise process called homologous recombination fixes double-strand breaks in the S as well as the G2 stage of the cell cycle. BRCA1 and BRCA2 are essential for the functioning of homologous recombination. Improper homologous recombination occurs when these protein’s function is lost due to inherited gene mutations.
Numerous biological processes, including metabolism, carcinogenesis, cancer growth, cell death, and replicative immortality, are regulated by the poly (ADP-ribose)-polymerase (PARP) enzyme family [60].The most prevalent and well-studied member of the PARP family, PARP-1, was implicated in the eukaryotic cell’s base excision repair (BER) pathway to repair DNA single-strand breaks[61].Damage to DNA triggers PARP-1, causing itto cleave its substrate nicotinamide adenine dinucleotide (NAD+) and provide nuclear target proteins with ADP-ribose units to aid in DNA repair[62]. The PARP-1 inhibitors have the ability to specifically target tumour cells that have faulty BRCA-1/2 genes[63]. Thus,targeting PARP is likely to be a promising method to fight TNBC.
Miaojiachen et. al., in 2021,[64]a series of 2-(4-[4-acetylpiperazine-1carbonyl]phenyl) -1H-benzo[d]imidazole-4-carboxamide derivatives were successfully synthesised and analysed.
These compounds were assessed utilising the PARP assay kit and MTT method for their PARP-1 inhibitory activity and cellular inhibition against BRCA-1 deficient cells (MDA-MB-436) and wild cells (MCF-7).The findings showed that furan ring-substituted derivatives 6a-6d shown superior PARP-1 inhibitory activity when compared to other heterocyclic compounds.Both 6c (IC50 = 43.56 ± 0.69?M) and 6d (IC50 = 36.69 ± 0.83?M) showed good antiproliferation activity on MDA-MB-436 cells and no effect on MCF-7 cells which indicated that the 6c and 6d have high selectivity and targeting.
Compound 6c was shown to have the most potent inhibitory effects on the PARP-1 enzyme (IC50 = 0.023?M), nearly matching standard drug, Olaparib's levels of inhibition.The binding manner of compound 6c and PARP-1 was investigated using the molecular docking approach, which suggested that hydrogen bond formation was necessary for PARP-1 inhibitory action.
PEPTIDYL-PROLYL ISOMERASE (PIN1) ENZYME
Human Pin1 is a member of the family of peptidyl-prolyl cis-trans isomerase which binds to proteins containing phosphorylated Ser/Thr-Pro sites. The isomerization of the pSer/pThr-Pro peptide bond can control the function of these proteins in different pathways such as gene promoters, instructs transcription complexes towards specific gene expression profiles, immune response, germ cell development, regulation of cell growth, genotoxic and other stress responses. Pin1 can bind to estrogen receptor,it increases the DNA binding to estrogen receptor so that the upregulation of Pin1 leads to different cancers, particularly breast cancer. Therefore, Pin1 is considered an important target for designing novel anti-breast cancer drugs.
Samira Nashaat et. al.[65], synthesised novel class of benzimidazole derivatives conjugated with either a six- or five-membered heterocyclic rings. The synthesised substances were tested biologically against breast cancer cell lines (MCF-7), and the compounds 7a, 7b and 7c displayed high activity (IC50 values 8.7 ± 0.6µM, 11.7 ± 0.9µM and 31.3 ± 2.5µ respectively and IC50 value of standard drug doxorubicin is 6.72µM) were then tested against normal human lung fibroblasts (WI38), and the IC50 values were found to be 71.6 ± 4.0µM, 67.9 ± 3.6 µM and 45.7 ± 2.8 µM for 7a, 7b and 7c respectively (IC50 of standard drug doxorubicin = 6.7 ± 0.5 µM) demonstrating their safety.
Using tannic acid as a reference control, compounds 7a,7b and 7c—which have the most anti-breast cancer activity against the MCF-7 cell line—were chosen for the Pin1 inhibition assay.The outcomes showed that each of the substances under investigation had extremely potent pin1 inhibitory action with IC50 values 0.76 ± 0.04 µM,0.64 ± 0.03µM and 1.10 ± 0.06µM for 7a, 7b and 7c respectively (IC50 value of tannic acid is 0.53 ± 0.02 µM). Investigated ability of compound 7ato induce apoptosis and its impact on cell cycle progression.Good agreement was found between the reported biological results and the molecular modelling analysis including docking studies of the most active compounds, 7a,7b and 7c, against Pin1 inhibition.
CONCLUSION
Worldwide, breast cancer is the primary cause of cancer-related deaths among women. Based on the expression of growth factor and hormone receptors, it is divided into a few main molecular subtypes likeluminal A (ER+ and/or PR+, and HER2-), Luminal B (ER+ and/or PR+, and HER2+), HER2 (ER-, PR-, and HER2+) and triple-negative breast cancer (TNBC) (ER-, PR-, and HER2-). Significant progress in the search for novel medications to treat BC has been made in the last few years.With a better understanding of the physiological variability of breast cancer, more tailored and efficient treatment plans can be created. Benzimidazole is a heterocyclic system which contains an imidazole attached to benzene ring. Diverse pharmacological effects of this system have been studied. Benzimidazole derivatives are available in the market for the treatment of various diseases. This bicyclic ring system is well known for its anticancer effects. This review article analysed the anti-breast cancer effects of benzimidazole derivatives through targeting various receptors like Estrogen Receptor, Aromatase enzyme, HER2, EGFR, Poly-ADP Ribose Polymerase (PARP), peptidyl-prolyl isomerase (pin1) enzyme.Review of various articles revealed that benzimidazole derivatives have the ability to elicit anti breast cancer effect by targeting suitable receptors.
REFERENCES


Krishna Priya A. , Aravind A. , Airin Benny, Benzimidazole Derivatives As Promising Agents Targeting Breast Cancer, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 9, 1501-1517. https://doi.org/10.5281/zenodo.13857533
 10.5281/zenodo.13857533
10.5281/zenodo.13857533