
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shri Gajanan Maharaj Shikshan Prasarak Mandal’s Dnyanvilas College of pharmacy, Dudulgaon, Pune 411081
This review offers a succinct summary of advancements made in analytical methods used for pharmaceutical degradation and impurity profiling, encompassing both active pharmaceutical ingredients (APIs) and finished formulations. It highlights emerging practices in forced-degradation studies and impurity assessment, organized by publication year, chromatographic parameters (such as column selection), sample type, and elution mode (isocratic or gradient). The article delivers a targeted, in-depth update on a range of analytical techniques including modern hyphenated methods used to detect impurities and degradation products at trace levels across various pharmaceutical systems.[1]
The pharmaceutical industry’s core mission is to protect public health by ensuring that patients receive medications that are safe, effective, correctly dosed, and affordable. Consequently, drug safety and therapeutic efficacy serve as the fundamental pillars of pharmacotherapy. A medication’s safety profile is shaped not only by its inherent pharmacological or toxicological properties but also by any impurities present in the active ingredient or the final dosage form. Thus, the overall safety of a drug depends on both the intrinsic toxicity of the API and nature of the impurities it contains.[1],[2],[3].
A pharmaceutical product must maintain its stability over its entire shelf life, ensuring that its identity, potency, purity, and overall quality are preserved. This is accomplished through strict quality control at every stage of production from the raw materials to the final product as well as ongoing monitoring after the product has been released to the market.
ICH guidelines categorize impurities into four main groups: organic impurities (such as starting materials, intermediates, process-related by-products, and degradation products), inorganic impurities (including salts, catalysts, ligands, and heavy metals), other extraneous materials and residual solvents. While this classification helps define impurity types, it does not account for enantiomeric or chiral impurities species with identical molecular formulas and connectivity but different three-dimensional arrangements, which can produce markedly different pharmacological or toxicological effects.[2]
Forced degradation studies play a critical role in revealing potential degradation products, clarifying degradation pathways, assessing the inherent stability of drug substances, and supporting the validation of stability-indicating analytical methods. Regulatory authorities recommend including these one-time degradation studies in Phase 3 IND submissions, and NDAs must contain detailed information derived from forced degradation, including identified degradation products, kinetic behaviour, structural characterization, mass balance data, and drug peak purity assessments.[3]
These studies help determine degradation mechanisms of the API alone and within the finished formulation, detect the presence of polymorphic or enantiomeric substances. Therefore, forced degradation and impurity profiling represent essential components of both IND and NDA documentation.
While numerous textbooks and review articles have discussed degradation and impurity studies, there is still a shortage of up-to-date literature that reflects current analytical advancements in this field. To fill this gap, the current review compiles recent advances in the analytical study of forced degradation and impurity profiling of APIs and pharmaceutical dosage forms. Research articles published between 2008 and 2012 were carefully analyzed, taking into account factors such as the drug matrix, therapeutic category, type of impurity or degradation product, chromatographic column properties, mobile-phase composition, elution technique, detection wavelength, and publication year, with the aim of identifying prevailing analytical trends.
Sources/ Types of Impurities in Medicines:
Pharmaceutical products must remain free of harmful or toxic impurities, and pharmacopoeias define permissible limits for these contaminants. Typical impurities that may occur in drug preparations include:[4]
|
Impurity Type |
Examples |
|
Crystallization-related impurities |
Polymorphism, Solvatomorphism |
|
Stereochemistry-related impurities |
Levaquin, Xooenex, Prilosec |
|
Organic Volatile impurities |
Benzene, tetrachlormethane, |
|
Starting Material and by-products |
GC-MS profiled impurities in ecstasy and MDMA from reductive amination. GC-MS analyzed impurities in MDMA and related intermediates. Impurities in ecstasy/MDMA were examined by GC-MS. |
|
Formulation-related impurities |
Microbial contamination can develop during a product’s shelf life and use. Multiple-dose products may become contaminated over time and with use. Contamination may occur both during storage and consumer handling. |
|
Impurities cause between storage |
Impurities can form during drug product storage or transport. Storage and shipping can generate various drug impurities. |
|
Method related impurities |
1-(2,6-dichlorophenyl) indolinone |
|
Mutual interaction amongst ingredients |
Nicotinamide, thiamine pyridoxine, riboflavin |
|
Functional group-related typical degradation |
Aspirin, benzocaine, cefotaxime |
|
Process-related drug substances |
Carbon-based compound, Raw material, Process intermediate, Side product, Contaminant in raw material |
|
Degradation drug product |
Organic products, Degradation products. |
|
Breakdown of the drug substance or medication |
Organic, Excipient interaction |
Raw Materials Used in the Manufacture of Pharmaceutical Substances:[7],[8],[9].
Pharmaceutical ingredients can originate from natural sources or be produced synthetically from chemical precursors. Natural sources include minerals, plants, animals, and microorganisms. Verifying the identity and quality of these raw materials is crucial, as any impurities present at the source may carry through the manufacturing process and ultimately cause in final drug substance. Minerals particularly are rarely found in a pure state; instead, they typically occur as mixtures containing chemically related substances. Examples include:
Systemic approach for Impurity determination
1. Method of Manufacture
The manufacturing process can introduce impurities into a pharmaceutical product, originating from reagents, catalysts, solvents, intermediates, or production equipment.
a. Reagents in Manufacturing:
Reagents may leave residual contaminants in the final product. For instance, calcium carbonate can retain small amounts of soluble alkali from the sodium carbonate used in its production, as traces may remain even after washing.
CaCl? + Na?CO? → CaCO?↓ + 2 NaCl
Another examples include:
b. Reagents Used to Remove Impurities
Sometimes, the very substances added to eliminate impurities become contaminants themselves. For instance, barium is used during potassium bromide production to remove sulphate residues, yet small amounts of barium may remain in the final product.
c. Solvents
Many pharmaceuticals crystallize in solvated forms, leaving behind solvent residues from synthesis, purification, or final crystallization.
Water, the most widely used solvent in inorganic pharmaceutical processes, can vary significantly in purity and therefore become a major contamination source. Types of water used include:
d. Reaction Vessels
Manufacturing equipment made from metals (e.g., copper, iron, stainless steel, silver, aluminium, nickel, zinc, lead) or glass can introduce impurities when corroded or chemically attacked by reagents.
Examples include:
e. Intermediates
Incomplete conversion of reaction intermediates can contaminate the final product.
For example, in the production of sodium bromide:
If the reduction step is incomplete, sodium bromate remains as an impurity.
f. Atmospheric Contamination
Air can carry dust (aluminium oxide, silica, soot, sulphur) and gases such as CO?, SO?, hydrogen sulphide, or arsine, all of which may contaminate products.
g. Manufacturing Hazards
Even when impurity controls are in place, certain risks remain during production:
I. Particulate Contamination:
Foreign particles such as dust, metal fragments, glass, plastics, or porcelain may enter the product from equipment wear or damaged containers. Eye ointments packaged in metal tubes, for example, may contain metallic particles.
ii. Cross-Contamination:
Airborne powder or tablet dust from one product can contaminate others manufactured in the same facility. This is especially dangerous with potent drugs like steroids or synthetic hormones.
iii. Microbial Contamination:
Liquid and topical formulations are prone to microbial entry during processing. Parenteral and ophthalmic preparations require sterility testing, and preservatives are used to minimize microbial growth.
2. Instability of the Product
Impurities may form in storage as a result of chemical degradation. Many drugs break down when exposed to unfavourable conditions. Factors such as light, residual acids or bases, metal ions, atmospheric oxygen, carbon dioxide, and moisture can accelerate decomposition. Understanding a drug’s chemical behaviour helps determine appropriate storage precautions.
b. Changes in Physical Properties
Physical instability may develop during storage, causing changes like crystal shifts, sedimentation, agglomeration, or caking, which can alter the product’s appearance, performance, and therapeutic effect.
c. Interaction with Container Materials
Packaging materials can introduce impurities by interacting with the product.
d. Temperature
Storage temperature significantly affects the rate of both chemical degradation and physical instability. Therefore, heat-sensitive products are assigned specific temperature requirements to minimize degradation.[12],[13].
3. Crystallization-related impurities [8],[9]
Studies show that a compound’s crystal form can strongly affect its solid-state properties, so regulators require close control of polymorphism and solvatomorphism.
4. Stereochemistry-Related Impurities
impurities can also result from stereochemical variations—molecules with the same formula but different 3D structures. Chiral compounds especially may show very different biological effects. Using a single enantiomer of a chiral drug is often advantageous because it may offer:
However, this is not always the case; for example, levofloxacin (the S-isomer) and ofloxacin (the R-isomer) display comparable pharmacokinetic behaviour. Examples of marketed single-enantiomer chiral drugs are levofloxacin, levalbuterol, and esomeprazole.[15].
5. Organic Volatile impurities
Organic Volatile impurities are volatile organic substances that may remain in drug materials after processing. Because some solvents pose toxicity risks, they must be minimized or eliminated during bulk drug manufacturing. According to ICH guidelines, these solvents are classified based on their potential harm to human health.[10].[16].
|
Class |
Description |
Examples |
Limit / Exposure |
|
Class I |
Highly toxic — should be avoided |
cyclohexa-1,3,5-triene, tetrachloromethane, dichloromethane, methyl alcohol, azabenzene, methylbenzene |
Very low ppm limits |
|
Class II |
Limited toxicity — restricted use |
N,N-dimethylformamide, acetonitrile |
Specific ppm limits |
|
Class III |
Low toxicity — safer |
Acetic acid, ethanol, acetone |
Allowed up to 50 mg/day |
A specialized GC method can assess the purity of solvents like acetone, dichloromethane, methanol, and toluene, while simultaneously quantifying impurities such as ethanol, isopropanol, chloroform, and benzene using propionitrile as an internal standard [11].[17].
6. Synthetic Intermediates and By-Products
Impurities in pharmaceutical compounds and new chemical entities (NCEs) can originate from the manufacturing process, such as from raw materials, reaction intermediates, or by-products formed during synthesis.[18].
7. Formulation-Related Impurities
solutions and suspensions are particularly prone to hydrolysis and solvolysis. For instance, Fluocinonide Topical Solution USP, 0.05%, was recalled in the U.S. due to the formation of degradation products that decreased its potency. Liquid dosage forms are particularly prone to chemical breakdown and microbial contamination. Key factors influencing impurity formation include:
Warm, humid conditions promote bacterial, fungal, and yeast growth, risking contamination in liquid products during storage or use, especially if preservatives are insufficient or packaging is permeable.[19],[20],[21].
8. Storage-Related Impurities
Impurities may also form during storage or transportation. Stability studies are therefore critical to ensure product safety and integrity throughout its shelf life.[22].
9. Method-Related Impurities
In diclofenac sodium parenteral formulations, autoclaving at 123?±?2°C causes intramolecular cyclization, forming 1-(2,6-dichlorophenyl)indolin-2-one and sodium hydroxide, with the impurity level highly dependent on the initial solution ph..[22].
10. Mutual Interaction Among Ingredients
Some vitamins, like thiamine in B-complex injections, are inherently unstable and can degrade during storage. Nicotinamide accelerates thiamine breakdown, causing the product to become sub-standard within a year, even with stabilizers and storage at pH 2.8–4.0.[23].
11. Functional group-dependent degradation
Many drugs undergo degradation due to the reactivity of their functional groups:[24][25],[26],[27].
|
Type of Degradation |
Examples |
|
Hydrolysis of esters |
Acetylsalicylic acid, Ethyl 4-aminobenzoate, Ethyl 4-hydroxybenzoate, Cefpodoxime proxetil |
|
Degradation by water |
Benzylpenicillin, oxazepam, lincomycin |
|
Oxidative Breakdown |
Hydrocortisone, methotrexate, catecholamines, morphine, vitamin A, unsaturated fatty acids |
Identification of Impurities [5],[6].
Impurities in pharmaceutical substances can be detected using several approaches:
i. Reference Standard Method
This approach guarantees the correct qualification and control of reference standards during drug development. Reference standards act as benchmarks to assess manufacturing processes and product quality, ensuring the final drug is safe for patients. They are necessary not just for the API, but also for impurities, degradation products, starting materials, intermediates, and excipients, making them essential for accuracy, consistency, and safety in pharmaceuticals.[14].
ii. Spectroscopic Methods
iii. Separation Methods
Analytical techniques used to isolate impurities include:
In TLC, a mixture is applied to a polar silica plate and developed with a relatively nonpolar solvent. Compounds separate based on polarity—polar impurities stick to the silica and move slowly, while less polar substances travel faster. Proper solvent choice ensures clear separation, and spots can be visualized using UV light or chemical stains.
In Gas Chromatography (GC), a vaporized sample is injected into a stream of inert carrier gas, which transports it through a column containing a stationary phase. Compounds separate according to their interactions with the stationary phase, influenced by properties like boiling point and polarity. Each compound exits the column at a characteristic time (retention time) and appears as a peak on the chromatogram, allowing detection and quantification of impurities.
In HPLC, impurities are separated by fine-tuning the mobile phase (solvents), stationary phase (column), flow rate, temperature, and detector settings. Separation relies on differences in compound properties such as polarity, size, or charge. For complex mixtures, gradient elution is often used, and stability-indicating methods help resolve drug impurities. The goal is to achieve baseline separation (resolution ≥ 2.0) for accurate identification and quantification. Success depends on optimizing interactions between the sample components and the column’s packing material.
Capillary Electrophoresis (CE) separates impurities by exploiting differences in molecules’ charge, size, and shape. By adjusting the electric field, buffer pH, and adding modifiers like cyclodextrins or surfactants, compounds migrate at different speeds, allowing impurities to separate from the main component and appear as distinct peaks on the detector. This high-resolution method uses a narrow buffer-filled capillary to separate charged species based on electrophoretic mobility, making it effective for detecting trace impurities.
A supercritical fluid—commonly CO?—is used as the mobile phase along with organic modifiers and a stationary phase such as silica. Compounds are separated based on polarity and other properties as they pass through the column. By adjusting pressure, temperature, and the proportion of modifiers, optimal resolution can be achieved. SFC often provides faster and more environmentally friendly separations than HPLC, particularly for chiral or complex mixtures.
Hyphenated Techniques: Chromatography can be combined with spectroscopic detectors (e.g., GC–MS, LC–MS, LC–MS–MS) to enhance structural identification and accuracy.
iv. Isolation Methods
Impurities may sometimes need physical separation for detailed study. However, modern instruments such as MS, NMR, and hyphenated methods often allow direct characterization without isolation.
Chromatographic Reactor Concept: An analytical-scale chromatographic column can serve both as a reaction chamber and a separation device. For example, HPLC in a chromatographic-reactor setup has been employed to examine the hydrolysis kinetics of the prodrug fosaprepitant dimeglumine. Similar methods have also been used to isolate impurities in drugs such as loratadine, celecoxib, and amikacin.
Techniques for impurity isolation include: [43],[44],[45],[46].
v. Hyphenated/ Characterization Methods
Hyphenated techniques combine a separation method with a spectroscopic detector, allowing efficient detection, identification, and characterization of impurities, particularly in complex mixtures. Common systems include:
Benefits: Identifying and analyzing impurities and degradation products provides a clearer understanding of drug stability, safety, and quality control..
Advantages :
Analytical Method Development
In the development of new drugs, it is crucial to generate precise and dependable analytical data throughout various stages. The main steps include:
Example calculation for signal-to-noise ratio (USP):
S/N = UV peak-to-peak noise2 × height × scale?
SN=2×height×scaleUV peak-to-peak noise ?
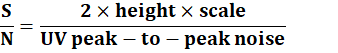
Residual solvents are categorized as follows:
Determining Limits:
Concentration (ppm)=(1000μg/mg)×PDE?dose
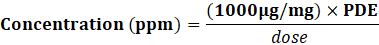
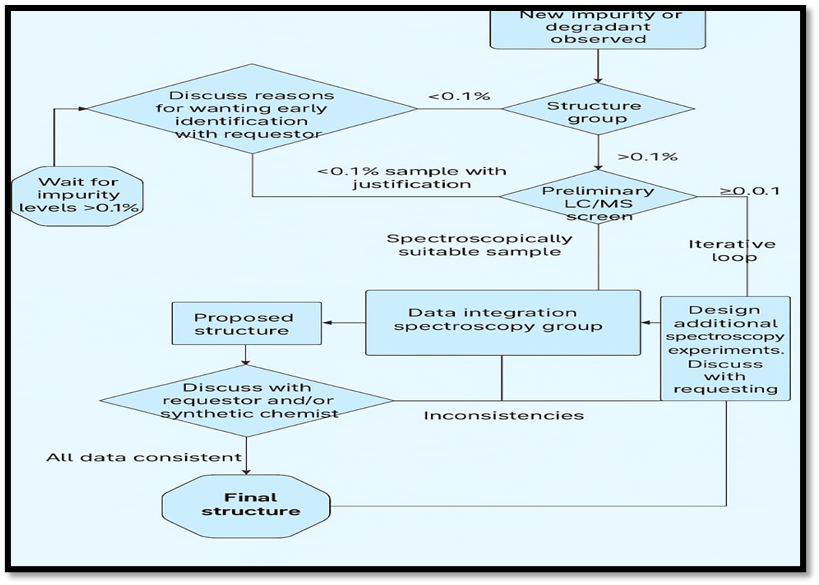
According to ICH guidelines, identification of impurities below 0.1% is generally not required unless they are unusually potent or toxic. For maximum daily dose:
Identification and Structural Elucidation of Impurities and Degradation Products
Conventional Approach: This method involves the separation, enrichment, isolation, or synthesis of impurities or degradation products, followed by spectral analysis. Common techniques include:
1. Solid-Phase Extraction (SPE):
2. Liquid-Liquid Extraction (LLE):
3. Supercritical Fluid Extraction (SFE):
CONCLUSION
This review highlights the pivotal importance of impurities in both drug substances and finished pharmaceutical products. Heightened public and media focus on drug safety has made impurity profiling a major concern in the pharmaceutical sector. The article examines the various types and classifications of impurities, approaches for their isolation and structural identification, analytical methods for their detection and quantification, and key factors to consider during bulk drug production.
In contemporary pharmaceutical practice, regulatory guidelines from various pharmacopoeias mandate the identification of impurities in Active Pharmaceutical Ingredients (APIs). Accurate isolation and structural analysis of these impurities are essential for obtaining safety information and evaluating their potential biological effects, highlighting the increasing importance of impurity profiling in pharmaceutical research and development.
REFERENCES


Rasika Tilekar, Smita Waghmare, Neelam Bhagdewani, Bhagyashri Gandhile, Vaibhav Ade, Pramod Ingale, Comprehensive Impurity Profiling and Degradation Pathway Elucidation Study, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 3073-3086. https://doi.org/10.5281/zenodo.18000273
 10.5281/zenodo.18000273
10.5281/zenodo.18000273