We use cookies to make sure that our website works properly, as well as some ‘optional’ cookies to personalise content and advertising, provide social media features and analyse how people use our site. Further information can be found in our Cookies policy
Congenital Insensitivity to Pain (CIP) is a rare hereditary sensory and autonomic neuropathy (HSAN) characterized by the inability to perceive pain while retaining other sensory modalities such as touch and temperature. Individuals with CIP often present with frequent injuries, burns, or bone fractures that go unnoticed due to the absence of pain perception. Mutations in genes such as SCN9A, NTRK1, and PRDM12 have been identified as major causes, leading to dysfunction in nociceptor development or sodium channel activity. Despite normal cognitive function, patients are at high risk of self-mutilation, joint deformities, and reduced life expectancy due to untreated injuries and infections. Early diagnosis, genetic counseling, and preventive care strategies are essential, as no definitive cure currently exists. Research on CIP not only helps in clinical management but also provides insights into pain mechanisms, offering potential therapeutic targets for novel analgesic drug development.
Congenital Insensitivity to Pain (CIP) is a rare and unusual genetic disorder characterized by an inability to feel physical pain. Although the condition sounds like it might be advantageous, it can lead to severe health risks, as individuals with CIP do not experience the warning signals that typically prompt people to avoid injury or seek medical attention for injuries. As a result, those with CIP can suffer from repeated undiagnosed injuries and complications that are often overlooked due to the lack of pain sensation.
CIP can be categorized into two distinct groups:2
Developmental disorders: In this group, nerve cells that detect pain (nociceptors) fail to develop or “die” early.
Functional disorders: In this group, nociceptors are present and correctly positioned in the body, but fail to respond to tissue-damage signals, leading to an inability to perceive pain.
CIP is caused by changes (variants) in different genes. Treatment is focused on the specific symptoms that the affected person has.
History of Congenital Insensitivity to Pain (CIP)
Congenital Insensitivity to Pain (CIP) has been a mysterious and rare condition that has fascinated medical professionals for centuries. The history of CIP spans a long journey from early observations of individuals who could not feel pain, to the genetic understanding of its underlying causes and mechanisms. Below is an overview of the key milestones in the history of CIP:
Early Observations and Case Reports
19th Century: First Medical Descriptions While the specific term CIP didn’t exist at the time, medical literature from the 19th century does include descriptions of individuals who were unable to feel pain. However, it wasn’t until the 20th century that physicians began to recognize the distinct nature of this condition.
One of the earliest recorded cases was in the 19th century when physicians described young children who would suffer severe injuries, such as broken bones or burns, without showing any signs of discomfort or distress.
These early cases were often met with confusion, as the idea of being unable to feel pain was a concept that didn't fit into medical understanding at the time.
Early 20th Century: Initial Research In the early 1900s, a few case reports began to gain attention, with some individuals exhibiting unusual behaviors, like self-mutilation or severe infections, that were later attributed to their inability to perceive pain.
However, without a proper understanding of the condition, these cases were often misinterpreted or confused with psychological disorders, especially because the inability to feel pain was such an extraordinary and puzzling phenomenon.
Mid-20th Century: Increasing Awareness
1950s–1970s: Recognition of the Condition The concept of congenital insensitivity to pain started to gain more recognition in the mid-20th century. In the 1950s, scientists began identifying specific features of the condition, but the genetic basis for CIP remained unclear.
Researchers observed that some families exhibited a pattern of autosomal recessive inheritance, suggesting that genetic mutations could be involved. This marked the beginning of efforts to understand the condition in a genetic context.
During this period, more cases of familial CIP were reported, especially in regions where close family relations (e.g., consanguinity) were common, which led to the theory that it might be a genetic disorder.
1980s–1990s: Genetic Advancements and Classification
Early Genetic Studies As genetic research advanced, especially with the discovery of DNA sequencing techniques, scientists started to investigate the genetic basis of CIP more rigorously.
In the 1980s, molecular genetics became a more prominent field, and researchers began to identify defects in specific genes that could be linked to the absence of pain sensation. Early genetic mapping techniques, although not fully advanced, helped point scientists in the direction of ion channel mutations.
1990s: Identification of the SCN9A Gene The major breakthrough in the history of CIP came in the 1990s, with the identification of the SCN9A gene. This gene codes for a sodium channel (Nav1.7) found in nociceptive (pain-sensing) neurons.
In 1994, researchers discovered that mutations in the SCN9A gene could lead to functional CIP, where the nociceptors (pain receptors) are present but do not function properly to transmit pain signals.
This discovery explained why some individuals had functional CIP, despite having nociceptors present, while others had the condition due to the absence of nociceptors (developmental CIP).
2000s: Advances in Genetic Research and Understanding
Molecular Mechanisms Unveiled The identification of the SCN9A gene and other related genes, such as PRDM12 and NGF, helped researchers understand the underlying molecular mechanisms of CIP more clearly.
With more genetic mapping and the advent of advanced genetic sequencing techniques, researchers began to identify specific mutations in these genes that prevent pain-sensing nerves from developing or functioning properly.
Case Studies and Medical Observations During the 2000s, more families with CIP were studied, leading to greater insight into the phenotypic variability of the disorder. For example, it was observed that some individuals with CIP might not only lack pain perception but also experience self-injurious behaviors (e.g., biting their tongue, fingers, or lips) or recurrent fractures due to their inability to sense injury.
2010s–Present: Ongoing Research and Therapeutic Approaches
Genetic Testing and Diagnosis Advances in genetic testing and next-generation sequencing now allow for the precise diagnosis of CIP in individuals, facilitating earlier detection and management of the disorder. The identification of specific mutations in genes like SCN9A and NGF has led to the development of genetic counseling tools for families at risk.
Discovery of New Genetic Mutations While SCN9A mutations remain the most common cause of functional CIP, other mutations have also been identified, such as those in the NGF (nerve growth factor) gene, which plays a crucial role in the development and survival of sensory neurons.
In 2010, a mutation in the NGF gene was linked to a developmental form of CIP, where the nociceptors did not develop properly in the first place, leading to congenital pain insensitivity.
Exploring Pain as a Therapeutic Target Research into CIP has also opened up potential avenues for understanding pain mechanisms in general. The study of SCN9A mutations has led to interest in developing pain-modulating therapies for people with chronic pain conditions. Some scientists are exploring how mutations in Nav1.7 (the sodium channel affected in CIP) can be targeted for the treatment of chronic pain in people who feel pain normally.
CIP and Quality of Life As understanding of CIP continues to grow, there is an increasing focus on improving the quality of life for those affected by the condition. Managing the increased risk of injury, infection, and joint damage is a key concern. Proactive care through regular checkups and educational programs for affected families are part of current approaches.
Signs and Symptoms of Congenital Insensitivity to Pain (CIP)
Congenital Insensitivity to Pain (CIP) is a rare and complex condition where individuals are unable to feel physical pain, but still experience other sensations like touch and temperature. While this may seem advantageous, it can result in significant health risks because the body’s natural defense mechanism (pain) is absent. Below is an overview of the key signs and symptoms of CIP:
1. Absence of Pain Perception
No pain sensation: The hallmark symptom of CIP is the complete lack of pain perception. Individuals with CIP do not feel pain, even in response to injuries such as burns, cuts, fractures, or bruises.
No pain from serious injuries: They may break bones, cut themselves, or burn their skin and not feel any discomfort or distress. Because of this, injuries often go unnoticed until they cause significant damage or complications.
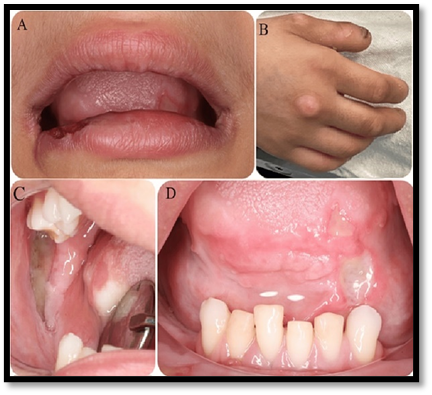
Fig.no1: Congenital Insensitivity to Pain with Anhidrosis
(A) ulceration and crusting in the lower lip
(B) biting marks on the fingers
(C, D) extensive ulcers in the retromolar pad and tongue, respectively.
2. Self-Injury and Unnoticed Injuries
Accidental self-harm: Due to the absence of pain, people with CIP are at high risk of unintentionally injuring themselves. Common incidents include biting their tongue, lips, or chewing their own fingers, leading to severe self-mutilation.
Unnoticed fractures: They may unknowingly break bones or sustain other significant injuries like dislocations and internal bleeding, which may not be detected for a long time.
Repetitive injuries: Without the sensation of pain, they might continue activities that can cause further harm (e.g., running on an injured foot or using a fractured limb). This results in recurrent injuries that often go unnoticed until later complications arise.
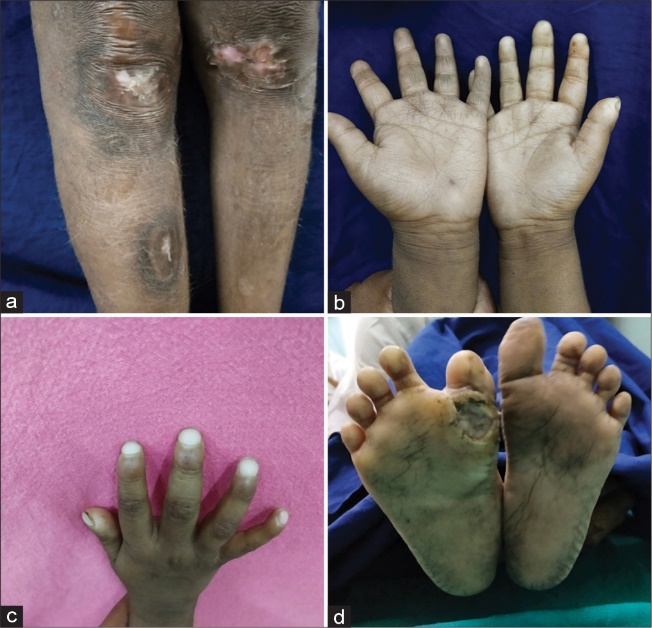
Fig.no2: Examples of common CIPA clinical presentations
3. Impaired Temperature Sensation
Difficulty sensing temperature extremes: Some individuals with CIP have an impaired ability to sense temperature, particularly extreme cold or heat. This can lead to:
Thermal burns: Due to an inability to feel heat, they may burn themselves without realizing it.
Frostbite: Similarly, exposure to cold temperatures may cause frostbite or hypothermia, as they don’t feel discomfort from cold.
Fig.No3: Temperature Sensation
4. Risk of Infections
Delayed detection of infections: Since individuals with CIP cannot feel pain, they may not recognize when they have cuts or wounds, leading to undiagnosed infections. These infections can spread unnoticed, and by the time they are discovered, they may be more serious.
Infected wounds or abscesses can become life-threatening if not promptly treated.
5. Joint and Bone Deformities
Repetitive joint injury: Without pain as a signal to stop, people with CIP may continue using damaged joints or limbs, leading to repetitive stress on the bones, ligaments, and joints.
Joint deformities can occur, especially in the hands, feet, and knees, due to undetected fractures or abnormal movement patterns.
Arthritis or other long-term joint complications may develop because the person doesn’t stop using an injured joint or bone.
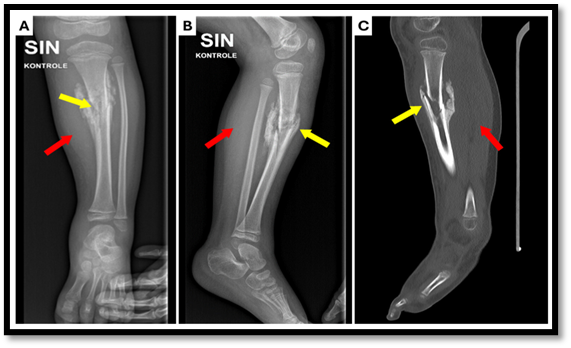
Fig. no4: (A) X-ray anterior–posterior projection
(B) X-ray lateral projection
(C) computed tomography (CT) scan of the lower left leg.
6. Delayed Wound Healing
Wound healing problems: When injuries occur (even minor ones), individuals with CIP may not notice if a wound is becoming infected or irritated, which can lead to delayed healing or chronic wounds.
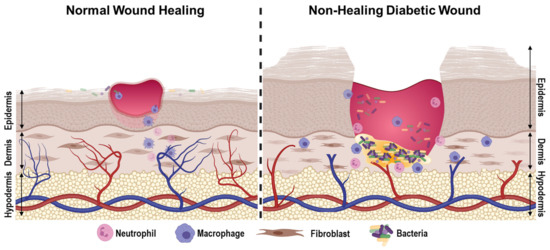
Fig No5: Diabetic Wound-Healing
7. Decreased Sweating and Heat Regulation (In Some Cases)
Autonomic dysfunction: Some individuals with CIP may also have a form of autonomic dysfunction, which affects their ability to regulate body functions like sweating, leading to an increased risk of overheating.
These individuals may not sweat normally, which can make it harder for them to cool down in hot environments.
8. Sensory Processing of Other Stimuli
Touch and pressure sensation intact: While people with CIP cannot feel pain, they typically retain the ability to feel other sensations like touch, pressure, and vibration. They may feel a light touch on their skin but will not feel any discomfort from sharp objects or damaging stimuli.
Proprioception: The sense of body position and movement (proprioception) is usually intact, which means individuals can sense where their limbs are in space and move normally, but without the pain cues to warn them against harmful movement.
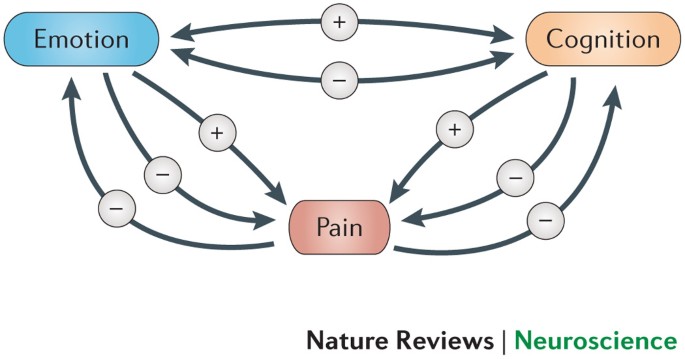
9. Neurological and Emotional Effects
Psychosocial impact: People with CIP, particularly children, may face emotional and social challenges due to their condition. Because they are unaware of the dangers of self-injury, they might be misunderstood or isolated.
They may also experience anxiety or depression due to the limitations on their social interactions and the constant vigilance required to avoid self-injury or harm.
Fig no6: Congnitive and Emotional Control of Pain
10. Cognitive Functioning and Development
Cognitive development: CIP typically does not affect a person’s intellectual or cognitive development. Most people with CIP have normal intelligence and can lead independent lives. However, intellectual development may be affected by injuries and infections that require long-term care or treatment.
Diagnosing a Congenital insensitive of pain
1. Clinical History
Start with a detailed patient history, especially in children.
Pain insensitivity: Patient does not feel pain (e.g., no reaction to burns, cuts, fractures).
Early onset: Symptoms usually appear in infancy or early childhood.
Self-mutilation: Common due to the inability to feel pain (e.g., biting tongue, fingers).
Recurrent injuries: Fractures, burns, bruises, or infections without clear cause.
No response to painful stimuli during immunizations or accidents.
Family history: May be autosomal recessive—consanguinity increases likelihood
2. Physical Examination
Look for scars, ulcers, orburn marks
Check for healing fractures, joint deformities, or osteomyelitis.
Inspect oral cavity and fingers/toes for signs of self-inflicted injuries.
Normal touch, temperature, and pressure sensations may be intact.
3. Neurological Assessment
Pain and temperature sensation are absent or significantly impaired.
Deep tendon reflexes may be present or absent, depending on subtype.
Cognitive development is often normal (but not always).
Motor function is usually preserved.
4. Diagnostic Tests
A. Quantitative Sensory Testing (QST)
Measures thresholds for temperature, pressure, and vibration.
Confirms selective loss of nociception (pain and temperature).
B. Nerve Conduction Studies / EMG
Often normal in CIP (distinguishes it from peripheral neuropathies).
C. Skin Biopsy
Intraepidermal nerve fibre density (IENFD): Reduced or absent small-diameter fibres (C-fibres).
Confirms small fibre neuropathy associated with CIP.
5. Genetic Testing
Essential for definitive diagnosis.
NGF (nerve growth factor) gene mutations
NTRK1 (TrkA receptor gene)
SCN9A (Nav1.7 sodium channel)
SCN11A, PRDM12, and others
Order a genetic panel for hereditary sensory and autonomic neuropathies (HSAN).
Differential Diagnosis
Exclude other conditions:
HSAN types I–V (CIP is part of this spectrum)
Acquired peripheral neuropathies
Autism (in cases with reduced response to pain)
Intellectual disability syndromes with self-injury
Psychological Assessment (Optional)
Assess for behavioural or developmental disorders if needed.
Important in children with self-injurious behaviour.
Dictional Formulation of Congenital Insensitivity to Pain (CIP)
Historically, Congenital Insensitivity to Pain (CIP) has been described as a rare neurological disorder in which individuals are unable to perceive physical pain due to genetic mutations affecting the pain-sensing system. The traditional understanding and formulation of CIP have focused on its clinical presentation, its genetic origins, and its impact on the nociceptive pathways (pain pathways) in the body.
1. Defining Features
The hallmark of CIP is the absence of pain perception in individuals who otherwise have normal sensory and motor functions. The traditional view recognized CIP as a disorder of the nociceptive system, which involves the detection, transmission, and processing of pain signals.
Key features traditionally emphasized in the formulation of CIP include:
Absence of pain: Individuals with CIP cannot feel physical pain, even in response to stimuli that would normally cause pain, such as burns, cuts, fractures, or internal injuries.
Normal sensation: Other forms of sensation, such as touch, temperature, and proprioception (body position awareness), remain intact. This means that individuals with CIP can still feel pressure, light touch, and temperature (heat and cold) but cannot feel pain.
Nociceptor dysfunction: The condition has traditionally been viewed as a disruption in the nociceptors—the specialized sensory receptors responsible for detecting painful stimuli. These nociceptors send pain signals to the central nervous system (CNS), but in CIP, this process is impaired or absent.
Autosomal recessive inheritance: The genetic basis of CIP is traditionally seen as autosomal recessive, meaning that the individual must inherit two copies of a defective gene (one from each parent) to manifest the disease.
2. Clinical Presentation
The classic clinical presentation of CIP involves the following:
No response to injury: Individuals do not react to injuries or painful stimuli. For example, they may sustain fractures, burns, or cuts without showing any signs of pain (e.g., crying, wincing).
Self-mutilation behaviors: Due to the lack of pain, individuals, especially children, may unintentionally bite their tongues or chew their lips. This often leads to self-inflicted wounds and damage to tissue.
Repetitive injuries: Since pain acts as a protective mechanism, individuals with CIP may continue activities that lead to further damage, such as walking on a fractured bone or using a burned hand.
Frequent infections: Lack of pain perception means that cuts, wounds, or abrasions may go unnoticed, leading to chronic infections and other complications like sepsis or delayed healing.
Thermal insensitivity: People with CIP may not feel heat or cold, making them more susceptible to burns or frostbite.
3. Pathophysiology (Nerve Pathways and Mechanism)
Traditional formulations of CIP focus on the dysfunction of nociceptors or pain receptors and their associated nerve pathways:ociceptors: These are specialized sensory neurons that detect painful stimuli such as mechanical injury, extreme temperature, or chemical irritants. They are a key component of the somatosensory system, which transmits pain signals from the body to the brain. In CIP, nociceptors either fail to develop properly or are present but cannot transmit pain signals effectively to the central nervous system.
Pathways to the brain: Once nociceptors are activated by a painful stimulus, they send signals to the spinal cord, which then transmits the signals to the brain. In individuals with CIP, there is a breakdown in this signal transmission due to genetic mutations.
The traditional theory was that the pain-transmitting pathways failed due to dysfunction at the level of:
Nerve development (e.g., defective or absent nociceptors).
Signal transmission (e.g., failure of ion channels involved in pain signal conduction).
4. Genetic Basis
The traditional formulation of CIP focused on the understanding that the condition was genetically inherited, most commonly in an autosomal recessive manner. In other words, both parents must carry a defective gene for their child to inherit the condition.
Mutations in specific genes: Over time, the discovery of the SCN9A gene, which codes for the Nav1.7 sodium channel (involved in pain signal transmission), became central to the understanding of CIP.
Mutations in SCN9A were identified as the most common cause of functional CIP.
This gene is crucial for the function of pain-sensing neurons, and mutations lead to dysfunction in nociceptors.
Another important gene identified in the traditional understanding of CIP is NGF (nerve growth factor), which plays a role in the development of sensory neurons.
Autosomal recessive inheritance: For an individual to develop CIP, they must inherit two defective copies of the gene (one from each parent). The parents of an individual with CIP are often asymptomatic carriers who each carry one copy of the defective gene.
Ayurvedic Supportive Treatment for CIP (Complementary Only): There is currently no scientifically validated Ayurvedic cure for Congenital Insensitivity to Pain (CIP), as it is a rare genetic neurological disorder. CIP is typically caused by mutations in genes like SCN9A, NTRK1, or NGF, which affect pain perception by altering nerve signalling pathways—this cannot be reversed or corrected by Ayurvedic medicine alone. However, Ayurveda can offer supportive care to help improve overall health, prevent complications, and support quality of life in CIP patients when used alongside conventional medicine.
A more structured approach doctors use to evaluate suspected CIP:
History & Clinical Signs
Absent or severely reduced reaction to painful stimuli from birth.
Self?injury (biting lips/tongue/hands), burns, fractures without complaints.
Possibly additional signs like anhidrosis (no sweating), temperature regulation problems.
Physical / Neurological Examination
Test pain stimuli (pinprick, heat) → no response. Check other sensory modalities (touch, temperature, proprioception) → often intact or variably affected.
Ancillary tests
Skin biopsy → to assess small nerve fiber pathology (loss of unmyelinated / small myelinated fibers) in some types.
Nerve conduction studies (often normal because large fibers often okay)
Genetic testing → to detect known pathogenic variants in one of the CIP?associated genes.
Differential diagnosis
Other neuropathies, acquired causes, abuse, trauma, etc.
Management / Follow?up
Once confirmed or strongly suspected, protective measures, frequent checks for injuries, treating complications.
Congenital Insensitivity to Pain (CIP) – Evolutionary Perspective
Congenital Insensitivity to Pain (CIP) is an extremely rare genetic condition, and from an evolutionary biology perspective, it presents a maladaptive trait, despite its fascinating molecular and neurological basis.
Pain: An Evolutionarily Conserved Protective Mechanism
Pain is one of the most critical evolutionary survival mechanisms. It:
Alerts organisms to injury or harmful stimuli.
Promotes withdrawal reflexes to avoid damage.
Prevents repeated harmful behaviors.
Encourages rest and healing.
CIP as a Result of Genetic Mutation
CIP arises from recessive mutations in key genes, including:
These mutations arerare and usually inherited in an autosomal recessive manner, meaning two defective alleles are needed for the condition to manifest.
Molecular Evolution of SCN9A and Related Genes
SCN9A gene:
Encodes Nav1.7 sodium channel, crucial for pain signaling.
Highly conserved in mammals, emphasizing its importance.
Mutations leading to loss of function → CIP
Gain of function mutations → extreme pain disorders (like primary erythromelalgia or paroxysmal extreme pain disorder)
Implications for Evolutionary Medicine & Pain Therapy
CIP has opened doors for pain drug development:
Targeting Nav1.7 pharmacologically to mimic CIP safely(pain relief without full insensitivity).
Studies on individuals with CIPhave informed evolutionary neuroscienceabout how pain circuits develop and adapt.
REFERENCE
Dearborn G. A case of congenital pure analgesia. J Nerv Ment Dis. 1932;75:612–5.
Rasmussen P. The congenital insensitivity-to-pain syndrome (analgesia congenita) Int J Paediatr Dent. 1996;6:117–22. doi: 10.1111/j.1365-263x.1996.tb00223.x.
Hall KH. Paediatric Orofacial Medicine and Pathology. 3rd ed. London: Chapman & Hall Medical; 1994. pp. 330–5.
Narayanan V. Oral and maxillofacial manifestations of hereditary sensory neuropathy. Br J Oral Maxillofac Surg. 1996;34:446–9. doi: 10.1016/s0266-4356(96)90105-9.
Nagasako EM, Oaklander AL, Dworkin RH. Congenital insensitivity to pain: An update. Pain. 2003;101:213–9. doi: 10.1016/S0304-3959(02)00482-7.
Dick PJ. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. In: Dick PJ, Thomas PK, Griffin JW, Low PA, Podreslo JC, editors. Peripheral Neuropathy. 17th ed. Philadelphia: Saunders; 1993. pp. 1065–93.
Emad MR, Raissi GR. Congenital insensitivity to pain with anhidrosis. Electromyogr Clin Neurophysiol. 2003;43:409–41.
Mardy S, Miura Y, Endo F, et al. Congenital Insensitivity to pain with anhidrosis (CIPA): effect of TRKA (NTRK1) missense mutations on autophosphorylation of the receptor tyrosine kinase for nerve growth factor. Hum Mol Genet. 2011;10(3):179–88. doi: 10.1093/hmg/10.3.179.
Kouvelas N, Terzoglou C. Congenital insensitivity to pain with anhidrosis. Ped Dent. 1989;11(1):47–51
Thompson CC, Park RI, Prescott Gh. Oral manifestation of the congenital insensitivity-to-pain syndrome. Oral Surg Oral Med Oral Pathol. 1980;50(3):220–5. doi: 10.1016/0030-4220(80)90373-4.
Littlewood SJ, Mitchell L. The dental problems and management of a patient suffering from congenital insensitivity to pain. Int J Paediatr Dent. 1998;8(1):47–50. doi: 10.1046/j.1365-263x.1998.00061.x.
Rosenberg S, Nagahashi Marie SK, Kliemann S. Congenital insensitivity to pain with anhidrosis (hereditary sensory and autonomic neuropathy type IV) Pediat Neurol. 1994;11(1):50–6. doi: 10.1016/0887-8994(94)90091-4.
Brahim JS, Roberts MW, McDonald H. Oral and maxillofacial complications associated with congenital sensory neuropathy with anhidrosis. J Oral Maxillofac Surg. 1987;45(4):331–4. doi: 10.1016/0278-2391(87)90354-5.
Shaaban H, Bayat A, Davenport P, et al. Necrotising fasciitis in an infant with congenital insensitivity to pain syndrome. British J Plast Surg. 2002;55(2):160–3. doi: 10.1054/bjps.2001.3771.
Auer-Grumbach M, De Jonghe P, Verhoeven K, et al. Autossomal dominant inherited neuropathies with prominent sensory loss and mutilations. Arch Neurol. 2003;60(3):329–32. doi: 10.1001/archneur.60.3.329.
Bonkowsky JL, Johnson CC, Smith AG, Swoboda KJ. An infant with primary tooth loss and palmar hyperkeratosis: a novel mutation in the NTRK1 gene causing congenital insensitivity to pain with anhidrosis. Pediatrics. 2003;112(3 pt 1):237–41. doi: 10.1542/peds.112.3.e237.
Rakocz M, Frand M, Brand N. Familial dysautonomia with Riga-Fede's disease: report of case. J Dent Child. 1987;54(1):57–9. ]
Nicholson GA, Dawkins JL, Blair IP, Kennerson ML, et al. The gene for hereditary sensory neuropathy type I (HSN-I) maps to chromosome 9q22.1-q22.3. Nat Genet. 1996;13(1):101–4. doi: 10.1038/ng0596-101.
Verhoeven K, Coen K, De Vriendt E, et al. SPTLC1 mutation in twin sisters with hereditary sensory neuropathy type 1. Neurology. 2004;62(6):1001–2. doi: 10.1212/01.wnl.0000115388.10828.5c]
Lafreniere RG, MacDonald M, Dube M, et al. Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the study of Canadian genetic isolates. Am J Hum Genet. 2004;74(5):1064–73. doi: 10.1086/420795. ]
Smael Labib, Berdai Mohamed Adnane, Abourazzak Sanae, Hida Mustapha, Harandou Mustapha. Congenital insensitivity to pain with anhydrosis: report of a family case. Pan Afr Med J. 2011;9:33. doi: 10.4314/pamj.v9i1.7120924. Congenital insensitivity to pain with anhidrosis Available. Access date: Nov 2010.
Reference
Dearborn G. A case of congenital pure analgesia. J Nerv Ment Dis. 1932;75:612–5.
Rasmussen P. The congenital insensitivity-to-pain syndrome (analgesia congenita) Int J Paediatr Dent. 1996;6:117–22. doi: 10.1111/j.1365-263x.1996.tb00223.x.
Hall KH. Paediatric Orofacial Medicine and Pathology. 3rd ed. London: Chapman & Hall Medical; 1994. pp. 330–5.
Narayanan V. Oral and maxillofacial manifestations of hereditary sensory neuropathy. Br J Oral Maxillofac Surg. 1996;34:446–9. doi: 10.1016/s0266-4356(96)90105-9.
Nagasako EM, Oaklander AL, Dworkin RH. Congenital insensitivity to pain: An update. Pain. 2003;101:213–9. doi: 10.1016/S0304-3959(02)00482-7.
Dick PJ. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. In: Dick PJ, Thomas PK, Griffin JW, Low PA, Podreslo JC, editors. Peripheral Neuropathy. 17th ed. Philadelphia: Saunders; 1993. pp. 1065–93.
Emad MR, Raissi GR. Congenital insensitivity to pain with anhidrosis. Electromyogr Clin Neurophysiol. 2003;43:409–41.
Mardy S, Miura Y, Endo F, et al. Congenital Insensitivity to pain with anhidrosis (CIPA): effect of TRKA (NTRK1) missense mutations on autophosphorylation of the receptor tyrosine kinase for nerve growth factor. Hum Mol Genet. 2011;10(3):179–88. doi: 10.1093/hmg/10.3.179.
Kouvelas N, Terzoglou C. Congenital insensitivity to pain with anhidrosis. Ped Dent. 1989;11(1):47–51
Thompson CC, Park RI, Prescott Gh. Oral manifestation of the congenital insensitivity-to-pain syndrome. Oral Surg Oral Med Oral Pathol. 1980;50(3):220–5. doi: 10.1016/0030-4220(80)90373-4.
Littlewood SJ, Mitchell L. The dental problems and management of a patient suffering from congenital insensitivity to pain. Int J Paediatr Dent. 1998;8(1):47–50. doi: 10.1046/j.1365-263x.1998.00061.x.
Rosenberg S, Nagahashi Marie SK, Kliemann S. Congenital insensitivity to pain with anhidrosis (hereditary sensory and autonomic neuropathy type IV) Pediat Neurol. 1994;11(1):50–6. doi: 10.1016/0887-8994(94)90091-4.
Brahim JS, Roberts MW, McDonald H. Oral and maxillofacial complications associated with congenital sensory neuropathy with anhidrosis. J Oral Maxillofac Surg. 1987;45(4):331–4. doi: 10.1016/0278-2391(87)90354-5.
Shaaban H, Bayat A, Davenport P, et al. Necrotising fasciitis in an infant with congenital insensitivity to pain syndrome. British J Plast Surg. 2002;55(2):160–3. doi: 10.1054/bjps.2001.3771.
Auer-Grumbach M, De Jonghe P, Verhoeven K, et al. Autossomal dominant inherited neuropathies with prominent sensory loss and mutilations. Arch Neurol. 2003;60(3):329–32. doi: 10.1001/archneur.60.3.329.
Bonkowsky JL, Johnson CC, Smith AG, Swoboda KJ. An infant with primary tooth loss and palmar hyperkeratosis: a novel mutation in the NTRK1 gene causing congenital insensitivity to pain with anhidrosis. Pediatrics. 2003;112(3 pt 1):237–41. doi: 10.1542/peds.112.3.e237.
Rakocz M, Frand M, Brand N. Familial dysautonomia with Riga-Fede's disease: report of case. J Dent Child. 1987;54(1):57–9. ]
Nicholson GA, Dawkins JL, Blair IP, Kennerson ML, et al. The gene for hereditary sensory neuropathy type I (HSN-I) maps to chromosome 9q22.1-q22.3. Nat Genet. 1996;13(1):101–4. doi: 10.1038/ng0596-101.
Verhoeven K, Coen K, De Vriendt E, et al. SPTLC1 mutation in twin sisters with hereditary sensory neuropathy type 1. Neurology. 2004;62(6):1001–2. doi: 10.1212/01.wnl.0000115388.10828.5c]
Lafreniere RG, MacDonald M, Dube M, et al. Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the study of Canadian genetic isolates. Am J Hum Genet. 2004;74(5):1064–73. doi: 10.1086/420795. ]
Smael Labib, Berdai Mohamed Adnane, Abourazzak Sanae, Hida Mustapha, Harandou Mustapha. Congenital insensitivity to pain with anhydrosis: report of a family case. Pan Afr Med J. 2011;9:33. doi: 10.4314/pamj.v9i1.7120924. Congenital insensitivity to pain with anhidrosis Available. Access date: Nov 2010.
Sachitanand Biradar
Corresponding author
Department of Pharmaceutics, Dayanand Institute of Pharmacy, Latur.






 10.5281/zenodo.17190927
10.5281/zenodo.17190927
