Department of pharmaceutics, Loknete Dr. J. D. Pawar College of Pharmacy, Manur (Kalwan) 423501, Maharashtra, India.
The poor aqueous solubility of many newly developed drugs, particularly those categorized as Biopharmaceutics Classification System (BCS) Class II, limits their oral bioavailability and therapeutic effectiveness. Liquisolid compact technology offers a promising approach to enhance the dissolution rate and bioavailability of poorly water-soluble drugs by converting liquid medications into dry, free-flowing, and compressible powders. In this study, a model BCS Class II drug was formulated as liquisolid tablets using microcrystalline cellulose (Avicel PH102) as the carrier, colloidal silicon dioxide (Aerosil 200) as the coating material, and polyethylene glycol 400 (PEG 400) as the non-volatile solvent. The liquid load factor was determined to ensure appropriate flow and compressibility of the formulation. The prepared liquisolid compacts were evaluated for pre-compression and post-compression parameters, including flow properties, hardness, friability, disintegration time, and in vitro dissolution behavior. The optimized formulation exhibited significantly improved dissolution profiles compared to conventional tablets, demonstrating the potential of liquisolid technology to overcome solubility-related challenges and improve oral drug delivery.
Oral administration remains the most popular route for drug delivery because of its convenience, safety, cost-effectiveness, and high patient compliance. Despite these advantages, many newly discovered drugs, particularly those belonging to Biopharmaceutical Classification System (BCS) Class II and IV, exhibit poor aqueous solubility, which leads to low and variable bioavailability1. This poses a major challenge for formulation scientists striving to achieve consistent therapeutic effects. Improving the solubility and dissolution rates of poorly water-soluble drugs has become a crucial goal in modern pharmaceutical development2. Several strategies have been developed to address solubility limitations, including particle size reduction (micronization and nanonization), solid dispersions, salt formation, complexation with cyclodextrins, self-emulsifying drug delivery systems (SEDDS), lipid-based formulations, and co-crystallization. Among these, the liquisolid compact technique has emerged as a promising and practical approach for enhancing the dissolution and bioavailability of water-insoluble drugs3. Liquisolid systems involve the transformation of liquid medications or drug solutions/suspensions into dry, free-flowing, and compressible powders suitable for tableting or encapsulation. This is achieved by incorporating the liquid drug into porous carrier materials and adsorbing excess liquid using fine coating materials, resulting in powders that look dry yet retain the drug in a solubilized or molecularly dispersed state. The core principle of liquisolid systems is based on the ability of carrier materials to retain liquid medication while maintaining acceptable flowability and compressibility4. The liquid load factor (Lf) is a crucial parameter and is defined as:
Lf = W/Q
Improved drug release from liquisolid systems is attributed to several factors: enhanced surface area available for dissolution, improved wettability due to hydrophilic liquid vehicles, and maintenance of the drug in a solubilized state without crystallization during compaction. Moreover, the dispersion of the drug at the molecular level in the carrier matrix contributes to rapid disintegration and dissolution, ultimately improving bioavailability5. The choice of non-volatile solvents is critical. Commonly used solvents include polyethylene glycol (PEG 200 and 400), propylene glycol, glycerin, polysorbate 80, and novel vehicles like Labrasol® and Capryol 90®. The solvent must provide high solubility for the drug and support the formation of a stable, compressible system. Carrier materials, such as microcrystalline cellulose (Avicel PH 102), lactose, mannitol, Neusilin®, and Sylysia®, play a vital role by absorbing the liquid medication and maintaining flow properties. Coating materials, like colloidal silicon dioxide (Aerosil®), silica gel (Syloid®), and magnesium aluminometasilicates, are used to cover the wet carrier particles, enhancing flowability and preventing sticking6. Liquisolid systems offer several advantages: they enable simple and scalable manufacturing without specialized equipment, are applicable to various poorly soluble and lipophilic drugs, improve dissolution and bioavailability, and represent a cost-effective alternative to complex technologies like solid lipid nanoparticles and self-emulsifying systems. However, they also have limitations, such as difficulty in handling high-dose drugs due to the large quantity of carrier and coating materials required, and potential compatibility issues between drug and excipients7. the liquisolid compact technique stands as a novel and effective approach for enhancing the dissolution rate and bioavailability of poorly water-soluble drugs. By transforming liquid medications into dry, free-flowing, and compressible powders using suitable carriers and coating materials, this technology overcomes solubility challenges and supports the development of robust oral solid dosage forms. The application of this technique can lead to better therapeutic outcomes, improved patient compliance, and potentially reduced dosing frequency, highlighting its significance in modern pharmaceutical formulation development8.
MATERIALS
Lumefantrine is used in the treatment of uncomplicated malaria caused by Plasmodium falciparum. The selected model drug was procured from a reliable pharmaceutical supplier. Excipients such as microcrystalline cellulose (Avicel PH102), colloidal silicon dioxide (Aerosil 200), polyethylene glycol 400 (PEG 400), and other required solvents and chemicals were obtained from certified suppliers and used as received. This Excipients used were of LR grade and sourced from Fine Chem Industries, Mumbai.
METHODS
Spectrometric and Compatibility Studies
a) UV spectroscopy was used to determine λmax and prepare the calibration curve.
b) FTIR analysis was performed to check drug-excipient compatibility.
c) DSC analysis was carried out to assess thermal behaviour and interactions.
Calculation of Liquid Load Factor (Lf)
Based on the solubility data and flowable liquid retention potential (Φ-value) of the carrier (Avicel PH102) and coating material (Aerosil 200), the liquid load factor (Lf) was calculated using the equation: where W is the weight of liquid medication and Q is the weight of carrier.
Preparation of Liquisolid Compacts
The drug was dissolved in the selected non-volatile solvent to prepare the liquid medication. The calculated amounts of carrier and coating materials were added gradually under continuous mixing to convert the liquid medication into a dry, free-flowing, and compressible powder. The powder blend was evaluated for flow properties (angle of repose, Carr’s index, Hausner’s ratio). The optimized liquisolid powder blend was compressed into tablets using a single punch tablet compression machine13.
Evaluation of Liquisolid Tablets
The prepared tablets were evaluated for various physicochemical parameters, including weight variation, hardness, thickness, friability, disintegration time, and drug content uniformity. In vitro dissolution studies were performed using USP type II paddle apparatus at 37 ± 0.5 °C in appropriate dissolution media. Samples were withdrawn at predetermined intervals and analysed spectrophotometrically to determine drug release profiles9,10.
In Vitro Dissolution Study
Dissolution testing was performed using a USP Type II (paddle) apparatus at 50 rpm and 37?±?0.5?°C in 900?mL of suitable medium (e.g., 0.1 N HCl or phosphate buffer pH 6.8). Samples were collected at specific intervals, filtered, and analyzed spectrophotometrically to determine drug release. Fresh medium was added to maintain volume. The cumulative percentage release was calculated and compared with conventional tablets11,12.
Stability Studies
Stability studies were conducted on optimized formulations according to ICH guidelines. Tablets were stored at 25 °C/75% RH for 6 months and analysed periodically for physical appearance, hardness, drug content, and in vitro dissolution.
Method of Preparation for Liquisolid Tablets
Table no. 1: Formulation Table
|
Sr. No |
Ingredients |
F1 |
F2 |
F3 |
F4 |
|
|
Lumefantrine |
20 |
20 |
20 |
20 |
|
|
Sesame oil |
0.3 |
0.3 |
0.3 |
0.3 |
|
|
MCC |
143 |
135 |
139 |
139 |
|
|
Aerosil 200 |
15 |
15 |
15 |
15 |
|
|
SSG |
4 |
8 |
8 |
4 |
|
|
Crospovidone |
4 |
8 |
4 |
8 |
|
|
PVP K30 |
10 |
10 |
10 |
10 |
|
|
Mg. Stearate |
4 |
4 |
4 |
4 |
|
Tablet weight |
200 |
200 |
200 |
200 |
|
Note: above formulation table all ingredient weight in mg, but Sesame oil is in ml
Table no. 4.2: Ratio and Liquid Load Factor for Formulation Table
|
|
Ratio (R) |
10:1 |
10:1 |
10:1 |
10:1 |
|
|
LF |
2 |
2 |
2 |
2 |
A The drug was first dissolved in a selected non-volatile solvent (e.g., PEG 400) to prepare the liquid medication. This liquid was then gradually mixed with the calculated amount of carrier material (MCC) under continuous stirring. After complete adsorption, the coating material (Aerosil 200) was added to improve flow properties and convert the mixture into a dry, free-flowing powder. The final blend was evaluated for flow characteristics and then compressed into tablets using a single-punch tablet press14,15.
Calculation for the W:
W= Drug + Non-volatile solvent
W = 20 + 0.32
W= 20.32
Liquid Load Factor (LF):
LF=WQ
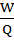
Where,
W= Weight of liquid blend
Q= Weight of carrier material
LF=20.3210
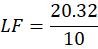
LF= 2.032
Non-Volatile solvent for the API
Solvent Volume=API (mg)Solubility (mg/ml)
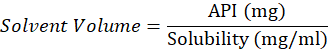
solvent Volume = 0.32 ml
Coating Material (q):
q=QR
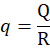
Where,
Q= Weight of carrier material
R= Ratio of Carrier material
q=1010
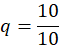
q = 1
Carrier (Q):
Q=WLF
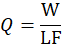
Where,
W= Weight of liquid blend
LF= Liquid Load Factor
Q=20.322.032
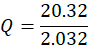
Q= 10
Ratio of the Tablet =
R value = carrier: coating
R=Qq
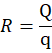
Where,
R= Ratio of Carrier
Q= Weight of carrier material
q = Weight of coating material
R=101
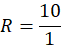
R= 10
RESULT
Determination of λ max of Lumefantrine
The determination of λ max of the Lumefantrine was performed in 0.1N HCL using by UV- Visible Spectrophotometer.
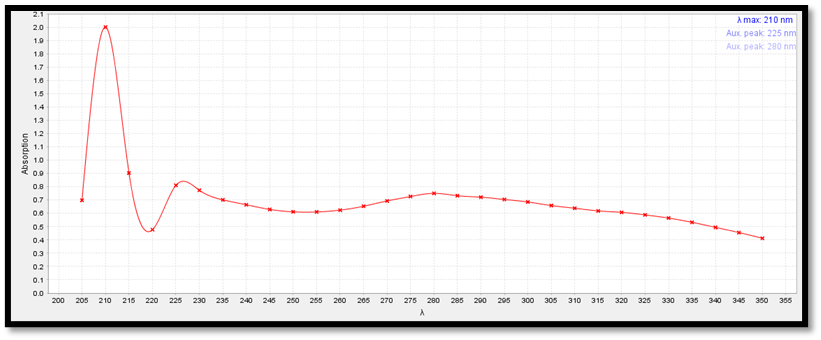
Fig 8.2 λ max of Lumefantrine in 0.1N HCL
Spectrometric Analysis
The calibration curve was obeyed Beer Lambert’s law in the concentration range of 0-50 µg/ml (R2 = 0.999).
Table no.2: Concentration and absorbance
|
Sr.No. |
Concentration |
Absorbance |
|
|
0 |
00 ± 00 |
|
|
5 |
0.1843±0.0025 |
|
|
10 |
0.2955±0.0047 |
|
|
15 |
0.4363±0.0064 |
|
|
20 |
0.5908±0.0078 |
|
|
25 |
0.7335±0.0090 |
|
|
30 |
0.8454±0.0133 |
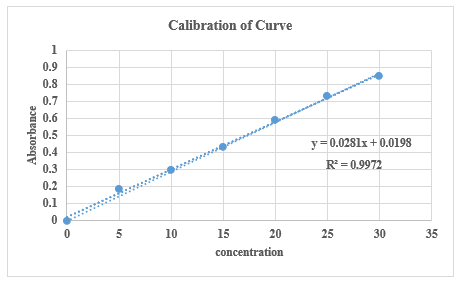
Fig. 1: Calibration Curve
Fourier transforms infrared (FTIR) spectroscopy
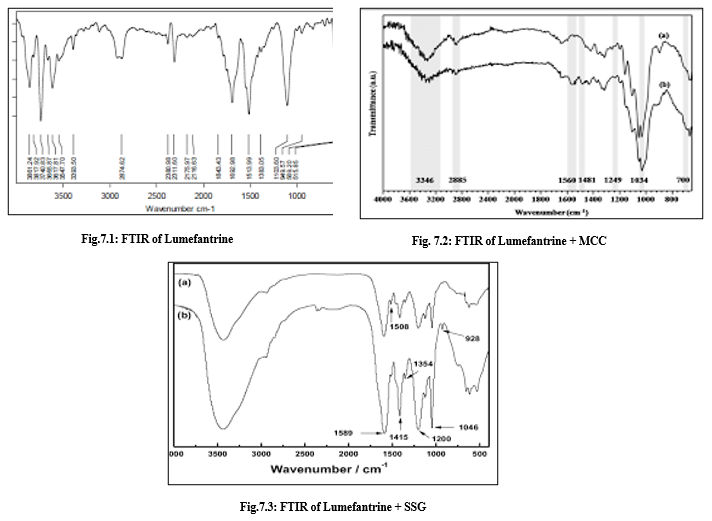
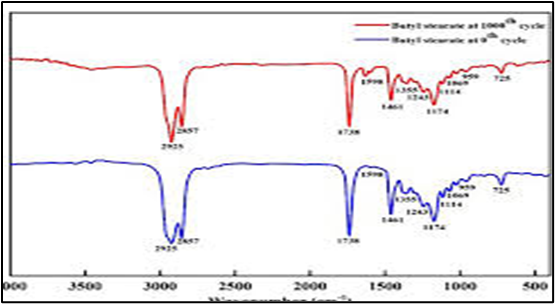
Fig.7.4: FTIR of Lumefantrine + Mg. Stearate
Discussion on FTIR Analysis
Fourier Transform Infrared (FTIR) spectroscopy was performed to assess any potential interactions between Lumefantrine and the excipients used in the liquisolid formulations. The characteristic peaks of pure Lumefantrine were compared with those of the physical mixtures containing various excipients. The FTIR spectrum of pure Lumefantrine (Fig. 7.1) showed characteristic peaks corresponding to its functional groups, including prominent absorption bands around 3340 cm?¹ (N-H stretching), 2920 cm?¹ (C-H stretching), 1640 cm?¹ (C=N stretching), and peaks in the fingerprint region confirming the integrity of the drug structure. FTIR analysis is a critical tool for evaluating drug-excipient compatibility in solid dosage forms. In this study, FTIR spectra confirmed that Lumefantrine maintained its characteristic peaks in all physical mixtures, indicating the absence of chemical interactions that could potentially affect drug stability or efficacy. This compatibility supports the safe incorporation of these excipients in liquisolid tablet formulations, ensuring that the drug remains stable during processing and storage. Thus, the FTIR results strongly support the feasibility of using these excipients for developing liquisolid compacts of Lumefantrine aimed at improving solubility and bioavailability.
Differential scanning colorimetry (DSC)
Fig.: DSC graph of Lumefantrine
DSC is a valuable technique for evaluating the physical state of the drug and for detecting potential interactions between the drug and excipients. In this study, the pure Lumefantrine exhibited a sharp melting endotherm, confirming its crystalline nature. However, in the liquisolid formulations, the decrease or broadening of this peak indicates a possible reduction in crystallinity. The absence of any new or unexpected peaks in the DSC thermograms of the formulations also confirms the absence of strong chemical interactions between Lumefantrine and the excipients. the DSC results complement the FTIR findings and further confirm that the liquisolid technique leads to a reduction in crystallinity of Lumefantrine, which is likely a key factor in enhancing its solubility and improving bioavailability.
Evaluation of Tablet
Table 6: Precompression evaluation of Tablet
|
Batch No |
Bulk density (gm/cm3) |
Tapped density (gm/cm3) |
Hausner’s ratio |
Carr’s index (%) |
Angle of repose (?) |
|
F1 |
0.383 |
0.444 |
1.18 |
14.61 |
27 °82 |
|
F2 |
0.379 |
0.448 |
1.18 |
15.46 |
28 °31 |
|
F3 |
0.378 |
0.450 |
1.16 |
15.41 |
28 °24 |
|
F4 |
0.377 |
0.446 |
1.13 |
12.14 |
29 °79 |
The flowability of powder blends is crucial for achieving good tablet uniformity and smooth processing. In this study, all formulations exhibited acceptable flow properties, Bulk density ranged from 0.377 to 0.383 g/cm³. Tapped density ranged from 0.440 to 0.450 g/cm³. Hausner’s ratio varied between 1.13 and 1.18. Carr’s index ranged from 12.14% to 15.46%. Angle of repose was found to be between 27°82′ and 29°79′. among the batches, formulations F2 showed slightly better flow properties.
Table 7: Post compression evaluation of tablet
|
Formulation |
Hardness (kg/cm2) |
Thickness (mm) |
Friability (%) |
Disintegration time |
% Assay |
|
F1 |
3 ±0.5 |
3.4±0.02 |
0.60±0.08 |
6.5±8 |
92.90 |
|
F2 |
3.5 ±0.2 |
3.4±0.02 |
0.23±0.2 |
6.2±4 |
95.76 |
|
F3 |
4 ±0.2 |
3.4±0.02 |
0.29±0.06 |
7.6±7 |
94.20 |
|
F4 |
4.5 ±0.5 |
3.5±0.02 |
0.40±0.05 |
6.4±5 |
93.04 |
The post-compression evaluation of liquisolid tablets (F1–F4) was conducted to assess their physical and mechanical properties of tablets. Hardness: All formulations exhibited sufficient mechanical strength to withstand handling and transportation. Notably, formulations F4 showed higher hardness values (4.5 kg/cm², respectively), which can be attributed to optimized carrier-to-coating ratios and better powder compatibility. Thickness: The slight variation in thickness among formulations indicates uniform die filling during compression. The consistent thickness reflects good flow and compressibility properties of the powder blends. Friability: All formulations showed friability well below the pharmacopeial limit of 1%, indicating excellent mechanical resistance. The lowest friability was observed in formulation F2, confirming its superior robustness.
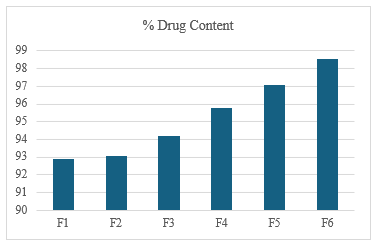
Drug content (assay): All batches showed uniform drug content within acceptable limits (90–99%), indicating good content uniformity. The highest assay value was observed in F6 (98.54%), suggesting efficient mixing and minimal loss during processing.
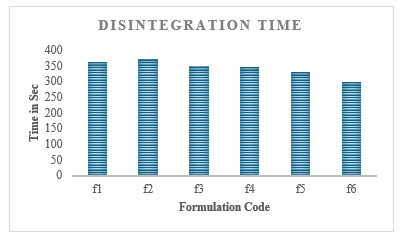
Disintegration time: Disintegration time is a critical factor affecting drug release and bioavailability. All formulations disintegrated within a reasonable time frame. F3 exhibited the fastest disintegration (7.6 min), likely due to its optimized formulation and higher super disintegrant content
In-vitro dissolution study
Table no. 7.9 In-Vitro Dissolution Study
|
Time |
F1 |
F2 |
F3 |
F4 |
|
0 |
0 |
0 |
0 |
0 |
|
5 |
19.5 |
26.25 |
25.5 |
21.25 |
|
10 |
42 |
46.5 |
43.5 |
40.75 |
|
15 |
70.5 |
78.25 |
74.25 |
69.12 |
|
30 |
84.29 |
96.42 |
90.21 |
88.77 |
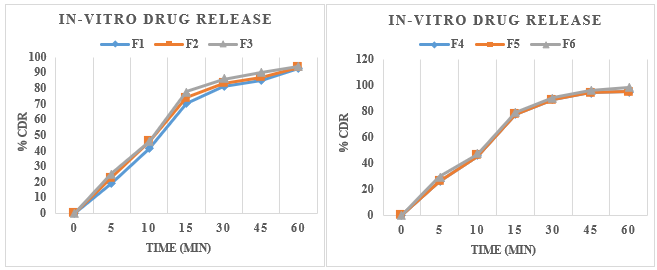
Fig. 7.8 % Drug Release
Optimization of formulation using 22 factorial designs
Equation in terms of coded factors
Drug Release= Final Equation in Terms of Coded Factors:
CDR = +89.92 -2.67 * A +0.43 * B -3.39 * A * B
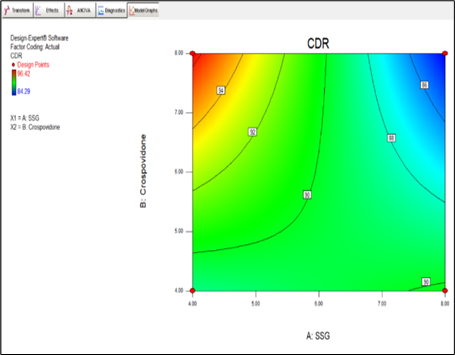
Fig.8.11 Contour plot for R1
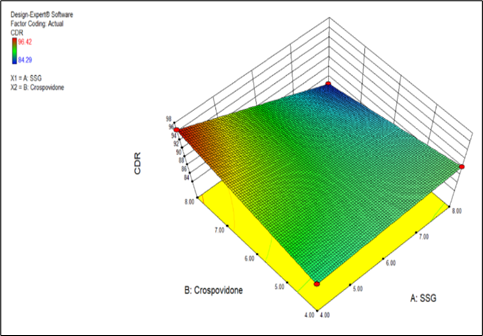
Fig 8.12 Effect of independent variables 3D surface plot
Table Analysis of variance for response R1 (% Drug release)
|
Source |
Sum of squares |
Df |
Mean square |
F value |
P value Prob > F |
Significance |
|
Model |
75.35 |
3 |
25.12 |
8.37 |
0.0422 |
Significant |
|
A- SSG |
28.57 |
1 |
28.57 |
4.33 |
0.1465 |
|
|
B- Crospovidone |
0.75 |
1 |
0.75 |
4.68 |
0.1347 |
|
|
AB |
46.04 |
1 |
46.04 |
|
|
|
|
Pure Error |
0.00 |
0 |
|
|
|
|
|
Cor total |
29.14 |
3 |
- |
|
|
Final Equation in Terms of Actual Factors:
CDR = +66.11000 +3.75250 * SSG +5.30500 * Crospovidone -0.84812 * SSG * Crospovidone
Final Equation in Terms of Coded Factors:
Friability = +0.38 +0.065 * A +0.035 * B +0.12 * A * B
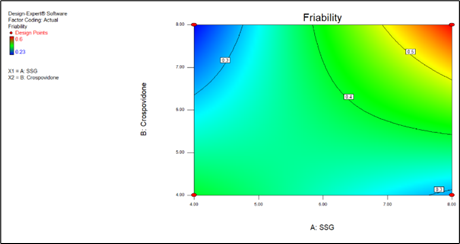
Fig.8.11 Contour plot for R2
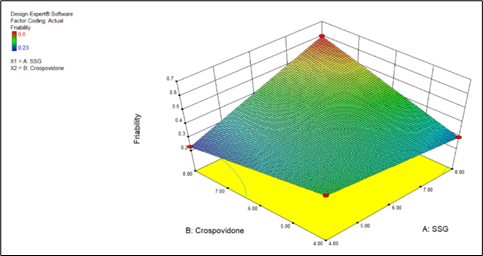
Fig 8.12 Effect of independent variables 3D surface plot
Table Analysis of variance for response R1 (Friability)
|
Source |
Sum of squares |
Df |
Mean square |
F value |
P value Prob > F |
Significance |
|
Model |
0.079 |
3 |
0.079 |
4.57 |
0.1154 |
Significant |
|
A- SSG |
0.017 |
1 |
0.017 |
2.41 |
0.1455 |
|
|
B- Crospovidone |
4.900 E-003 |
1 |
4.900 E-003 |
1.22 |
0.1325 |
|
|
AB |
0.058 |
1 |
0.058 |
|
|
|
|
Pure Error |
0.000 |
0 |
|
|
|
|
|
Cor total |
0.079 |
3 |
- |
|
|
Final Equation in Terms of Actual Factors:
Friability = +1.16000 -0.14750 * SSG -0.16250 * Crospovidone +0.030000 * SSG * Crospovidone
CONCLUSION
The present study demonstrated that liquisolid compact technology is an effective approach to enhance the dissolution rate and oral bioavailability of poorly water-soluble drugs. By converting the drug into a liquisolid system using a suitable non-volatile solvent, carrier, and coating material, the prepared tablets showed significantly improved dissolution profiles compared to conventional formulations. This technique offers a simple, cost-effective, and scalable strategy to overcome solubility-related challenges, making it a promising tool in the development of solid oral dosage forms.
Conflict Of Interest
Regarding this study, the authors report that they have no conflicts of interest.
ACKNOWLEDGMENTS
The author expresses heartfelt gratitude to Loknete Dr. J. D. Pawar College of Pharmacy, Department of Pharmaceutics, Manur, Tal. Kalwan, Dist. Nashik, for providing the necessary facilities and support to carry out this research work. The author is deeply thankful to Mr. Yeshpal More Sir, for his valuable guidance, constant encouragement, and insightful suggestions throughout the study. The author also extends sincere thanks to all faculty members, friends, and family for their continuous support and motivation.
REFERENCES

Vaibhav Patil*, Yashpal More, Formulation and Evaluation of Liquisolid Tablets of Lumefantrine for Enhanced Solubility and Bioavailability, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 604-616. https://doi.org/10.5281/zenodo.16751734
 10.5281/zenodo.16751734
10.5281/zenodo.16751734
