
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Samarth Institute of Pharmacy Belhe, Pune, Maharashtra, India.
Effective documentation control is a cornerstone of Good Manufacturing Practices (GMP) and is a regulatory imperative across pharmaceutical and healthcare industries. This review explores key principles of Good Documentation Practices (GDP), detailing the essential components of document creation, management, and maintenance. Core areas include proper formatting, accurate record-keeping, handling of electronic documentation, error correction protocols, and long-term data preservation. High-quality documentation reflects the operational integrity and compliance standards of a pharmaceutical organization. The paper emphasizes that whether maintained on paper or electronically, documentation must uphold characteristics such as being attributable, legible, traceable, permanent, contemporaneous, original, and accurate. Robust documentation not only supports the development, approval, and lifecycle management of pharmaceutical products but also plays a pivotal role in ensuring data reliability and regulatory transparency. Recommendations provided aim to strengthen site-level documentation systems, fostering a culture of quality and accountability.
Good Manufacturing Practices (GMP) are fundamental to the pharmaceutical and healthcare sectors, where the accuracy and reliability of documentation significantly influence product quality and regulatory compliance. Documentation is not merely a procedural formality it is a legal and ethical obligation that underpins traceability, consistency, and accountability across all stages of product development and lifecycle management.
GMP requires structured documentation systems that include specifications, manufacturing and packaging instructions, batch records, and validation protocols. These records are essential for demonstrating compliance with regulatory standards, and they allow for the traceability of each batch for a defined retention period commonly at least one year beyond expiration. Such documentation helps ensure that every product is manufactured and controlled according to standards appropriate for its intended use.[1]
Good Documentation Practices (GDP), as defined by the World Health Organization (WHO), are critical components of quality assurance. GDP encompasses all methods of generating, maintaining, correcting, and securing data to safeguard its integrity throughout a product's lifecycle. Whether documents are handwritten or electronic, they must be attributable, legible, traceable, permanent, contemporaneously recorded, original, and accurate.[2]
Clearly structured and verified documents help prevent errors, particularly in operations such as formulation, testing, packaging, and record-keeping. Unlike verbal communication, which can be ambiguous and unverifiable, written procedures like Standard Operating Procedures (SOPs), Master Formula Records, and batch documentation create a definitive audit trail.
Regulatory authorities like the U.S. FDA have codified expectations for documentation through frameworks like 21 CFR 211.180(e), which emphasize the necessity of written records for assessing drug quality. Compliance with these standards is mandatory for pharmaceutical companies and their collaborators, and non-compliance can lead to regulatory actions.
This paper presents an in-depth review of GDP, with focus areas including:
Through this exploration, the aim is to underscore the role of GDP in building a reliable and compliant quality management system one that safeguards both product integrity and patient safety.[1-2]
Principles of Good Documentation Practices (GDP)
Good Documentation Practices (GDP) form the foundation of quality assurance and are integral to compliance with Good Manufacturing Practices (GMP). These principles govern the creation, control, and preservation of documents that reflect the manufacturing, testing, and distribution activities of medicinal products.
Documentation in pharmaceutical environments may take diverse forms including handwritten records, electronic systems, and photographic media. Regardless of format, all documentation should be clearly defined within the organization’s Quality Management System (QMS), which must outline standards for content, structure, approval, retention, and disposal.[3]
The primary objectives of GDP are to:
To achieve these goals, the QMS should provide:
There are two principal categories of documentation:
Effective GDP also requires:
At its core, GDP ensures that all documentation whether physical or electronic is secure, attributable, accurate, contemporaneous, and traceable, thereby upholding product quality and regulatory compliance.[4]
Types of Documents
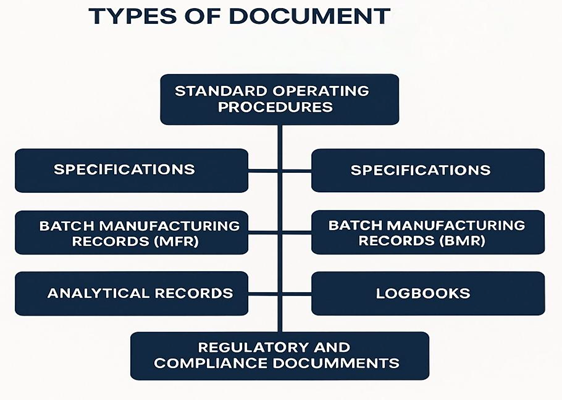
Figure 1: Types of Documents
Documentation Lifecycle
The documentation lifecycle encompasses creation, review, approval, distribution, storage, revision, and archiving. Each stage ensures traceability, compliance, and accountability. Control mechanisms must be in place to maintain data integrity throughout the lifecycle.[6]
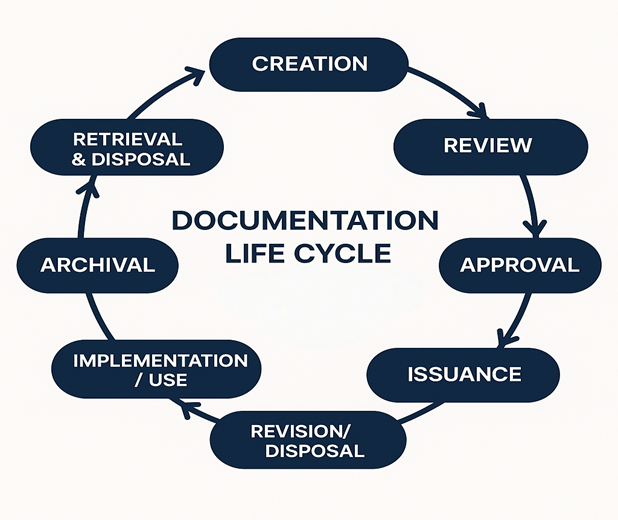
Figure 2: Documentation Life Cycle
Key Elements of Good Documentation Practice (GDP)
GDP relies on principles that make records accurate, legible, timely, and traceable. Essential elements include:
Electronic Documentation and Data Integrity
With the rise of digital platforms, electronic documentation is governed by data integrity principles such as ALCOA+:
Systems must validate electronic records for authenticity, security, audit trails, and backup protocols.[8]
Common Documentation Errors and Deviations
Errors frequently cited by regulatory bodies include:
Training and Awareness for GDP Compliance
Personnel must undergo regular training to ensure understanding of GDP principles. Key focus areas:
GDP in Regulatory Audits and Inspections[10]
Inspectors evaluate GDP as part of GMP audits. Areas of scrutiny include:
Challenges in Implementation of GDP
Organizations face several hurdles:
Recent Trends and Innovations in Documentation
Innovative developments include:
CONCLUSION
This review has underscored the critical importance of good documentation practices (GDP) within pharmaceutical quality assurance systems. Each document type from specifications to batch records not only reflects regulatory requirements but also plays a vital role in building a transparent, traceable, and controlled manufacturing environment. GDP ensures that all procedures, decisions, and quality standards are faithfully recorded, facilitating regulatory audits and supporting the integrity of product development and lifecycle management.
Effective documentation is far more than recordkeeping; it is a strategic quality tool that safeguards compliance with Good Manufacturing Practice (GMP), strengthens data reliability, and enables continuous improvement. By integrating GDP principles into the daily operations of pharmaceutical organizations through structured procedures, digital systems, training programs, and routine document reviews companies can foster a culture of accountability and excellence.
In essence, the consistent application of GDP transforms documentation into a powerful framework for regulatory confidence, patient safety, and sustainable product quality.
REFERENCES

Omkar Gosavi, Dr. Sachin Datkhile, Komal Aher, Divya Deshmukh, Sahil Patel, Bhakti Gurav, Komal Gaikwad, Good Documentation Practices (GDP) in Pharmaceutical Quality Assurances, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 669-675. https://doi.org/10.5281/zenodo.17063364
 10.5281/zenodo.17063364
10.5281/zenodo.17063364